

Definisjon
Marfans syndrom er en arvelig (genetisk) sykdom (autosomal dominant) forårsaket av defekt (misfolding) i proteinet fibrillin-1som kodes av genet FBN1 på kromosom15. Således er Marfans syndrom en medfødt, genetisk tilstand som skiller seg fra de betennelseslignende, systemiske autoimmune bindevevssykdommene. Omtrent 25% er spontanmutasjoner uten familiedisposisjon (referanse: Store Norske Leksikon, A Heiberg 2009). Sammen med en vaskulær form (som angriper blodårer) av Ehlers-Danlos syndrom, Loeys-Dietz syndrom og arvelig thorakalt aortaaneurisme (HTAD), inngår sykdommen blant de arvelige” Marfans-lignende” sykdommene.
Forekomst
Antall nye tilfelle hvert år (insidens) av Marfans syndrom er 2-3/10000. Forutsatt at alle erkjennes var tilsvarende 1/9802 nyfødte i en skotsk studie (referanse: Gray JR, 1994). Forekomsten i befolkningen (prevalens) er beregnet til : en blant 20.000 personer (ca. 500 i Norge). Marfans sykdom defineres dermed som en sjelden sykdom (færre enn 1 pr 2.000 personer i henhold til Det Norske Helse og Omsorgs-departementet). Likevel regnes den som den en av de vanligste arvelige sykdommer som angriper muskel-skjelettsystemet.
Symptomer
Skjelettsystemet
- Påfallende stor kroppshøyde (referanse: Erkula G, 2002).
- Skjev rygg (skoliose), lange, armer og ben (dolichostemomeli) med lange fingre og tær (arachnodaktyli) i forhold til kroppens lengde er vanlig (referanse: Lindsey J, 1998).
- Brystben deformiteter som fuglebryst (pectus carniatum) og overbevegelige (hypermobile) ledd ses også ofte (referanse: Kaissi AA, 2013).
Øyne
- Ofte foreligger alvorlige synsforstyrrelser med nærsynthet (myopi) og astigmatisme (skjeve hornhinner). Linse subluksasjon (ektoptisk linse) påvises av øyelege. Noen utvikler glaukom (grønn stær) eller tidlig katarakt (grå stær) (referanse: Zech J-C, 2020).
Hjertet og blodårer
- De mest alvorlige komplikasjoner som kan være livstruende og omfatter uregelmessig hjerte-rytme, hjerteklaff-lekkasje, utvidet diameter på hovedpulsåren (aortaaneurisme), aortadisseksjon (skade og lekkasje i blodåreveggen) (referanse: Rybczynski M, 2010).
- Spesiell oppmerksomhet i svangerskap er viktig.
Lunger. Spontan lungepunktering (pneumotoraks) forekommer, oftest utgående fra emfysem i lungenes overlapper (øvre del).
Hud. Strekkmerker (striae) skyldes hypermobilt bindevev.
Diagnosen
Mistanke om Marfans syndrom får en på bekgrunn av symptomer og undersøkelsesfunn (se ovenfor).
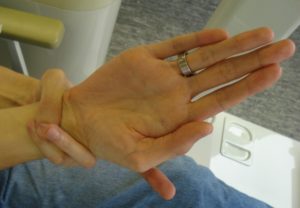
Ofte brukes også kriterier for diagnosesetting (2010 Ghent) som skiller mellom de som har Marfans sykdom i nærmeste familie og de uten arvelig disposisjon:
Uten arvelig disposisjon:
- Aortarot utvidet (Z-skår ≥2) og ektopisk linse
- Aortarot utvidet (Z-skår ≥2) og FBN1 mutasjon (gentest)
- Aortarot utvidet (Z-skår ≥2) og systemisk skåring* >7 punkter (spesielt skåringssystem*)
- Ektopisk linse og FBN1 mutasjon med kjent aortaaffeksjon
Med familie med Marfans sykdom:
- Ektoptisk linse
- Systemisk skåring* Aortarot utvidet ≥7
- Aortarot utvidet (Z-skår ≥2)
*Skår: Pectus carniatum=2 (pes excavatum eller assymmetri=1), Malleol-deformitet (ankel)=2 (plattfot=1), Dural ektasi=2, Protrusio acetabuli=2, Dys-proporsjonal armlengde =1, Skoliose eller kyfose=1, Redusert albue ekstensjon=1, Ansiktsskjelett forandringer= 1, Striae i huden=1, myopi=1, Mitralprolaps =1.
Gamle kriterier: major kriterier (minst 4):
- Redusert proporsjon overkropp/underkropp ratio (0,85 vs 0,93 hos normale)
- Arachnodactyli (fingre, tær), positiv tommel-håndledd-test)
- Skoliose mer enn 20 grader eller spondylolistese
- Medial malleol-feilstilling som gir pes planus (plattfot)
- Redusert ekstensjon i albuer (<170 grader)
- Pectum carniatum (fuglebryst) (Differensialdiagnose: Morquio syndrome, Noonan syndrome, Trisomi 18, Trisomi 21, Homocystinuri, Osteogenesis imperfecta, multiple lentigines syndrom, and Sly syndrome)
- Pectus excavatum (traktbryst) som krever kirurgi
- Protrusio Acetabuli (Differensial diagnoser: Pagets syndrom, revmatoid artritt, Bekhterevs sykdom, Osteomalasi)
Genetikk / arvelighet
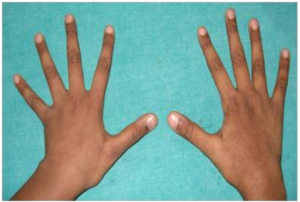
Ofte foreligger spontanmutasjon (25%), uten at tilsvarende sykdom ses i nærmeste slekt. Mutasjon i genet FBN1 kan påvises hos ca. 85% med klinisk sikker Marfans sykdom (referanse: Store Norske Leksikon, Heiberg A, 2009). Prøve kan sendes Oslo Universitetssykehus, Ullevål. Mer om genetisk tester på genetikkportalen.
Prognose
Det er ennå ingen behandling som kurerer Marfans syndrom. Men prognosen er bedret veldig mye de siste ti-år. Dette har sammenheng med nytte av regelmessig medisinsk oppfølging som inkluderer kontroller hos hjertespesialist (kardiolog), undersøkelser av blodårer for å utelukke eller følge opp aneurismer og behandling av pneumotoraks.
Lignende sykdommer/ differensialdiagnoser
En må ikke glemme at mange friske har flere av symptomene uten å være angrepet av sykdommen. Ved sterk mistanke om Marfans sykdom kan en i utredningen vurdere:
- Arvelig thorakalt aortaaneurisme (HTAD)
- Ehlers-Danlos syndrom
- Ektoptisk linse syndrom (ELS), familiær thorakalt aorta aneurisme og disseksjon syndrom (FTAAD/FTAA).
- Hypermobilitetssyndromet
- Loeys-Dietz syndrom (TGF-beta receptor 1 og 2 genmutasjoner, prøve sendes OUS, UUS)
- Menkels sykdom (defekt i kobber-metabolisme)
- Pseudoxanthoma elasticum
- Stickler syndrom (hereditær arthro-oftalmopati).
- Weill-Marchesani syndrom (WMS).
Behandling
Bruk av beta-blokkere som reduserer blodtrykket, regelmessige kontroller, begrenset tung fysisk aktivitet og operasjon av utposninger på hovedpulsåren og svikt i hjerteklaffer har bedret prognosen vesentlig, men ingen helbredende behandling er tilgjengelig. Regelmessig kontroll av blodårene er viktig. Gjennomsnittsalder for operasjon er 32 år. Stabiliserende ortoser og tilpasset fottøy. Behandlingen er ellers avhengig av eventuelle komplikasjoner fra øyne, ledd og columna / thoraks. Ingen helbredende (gen-terapi) behandling er tilgjengelig.
- Dersom utvidelse av pulsårer (aneurismer) øker, kan operasjon for å forebygge at pulsåren sprekker (aortaruptur) bli nødvendig.
- Kar-kirurger / thoraks-kirurger har retningslinjer for når operasjoner bør gjøres.
- Blodtrykksmålinger er viktige for å oppdage eventuelt høyt blodtrykk som kan øke risikoen for aortaaneurismer
- Vanlig fysisk aktivitet anbefales. Intensiv, hard fysisk trening øker trykket på blodårene og bør unngås
- Kortison og annen immundempende behandling har ingen plass i behandlingen av arvelige bindevevssykdommer, til forskjell fra autoimmune bindevevssykdommer
- Tabletter i form av en betablokker kan redusere blodårekomplikasjoner og anbefales for mange
- Behandlingen er ellers avhengig av eventuelle komplikasjoner fra øyne, ledd, virvelsøyle og tilsvarende
Kontroller kan omfatte MR-angiografi fremstiller de viktigste pulsårene fra og med hodet, halsen, brystet, mageområdet og bekkenet. Også med ultralyd Doppler / ekkokardiografi kan diameter på hovedpulsåren ved hjertet og aortabuen måles. Hvis pulsåren utvider seg, gjøres målinger minst hvert halvår, ellers årlig i en lengre periode. Også ved CT-undersøkelse kan hele hovedpulsåren fremstilles. Undersøkelsen gjøres ikke for ofte på grunn av røntgenstråling.
Litteratur
- Kompetansesenter, Sunnås Sykehus
- Salik I, 2022
- Dietz HC, 2001
- Marfan-foreningen i Norge
- Barstow C, 2015
- Grans Kompendium i Revmatologi