

Amyloidose (ICD-10 E85)
Definisjon
Amyloidose er gruppe sykdommer hvor spesielle proteiner (autologe fibrillære proteiner) klumper seg sammen i bindevevet utenfor cellene. Denne opphopningen av proteiner, kalt amyloid, fortrenger det normale vevet og skader etter hvert organfunksjonene. Det finnes flere typer amyloidose. En type, kalt AA-amyloidose (sekundær amyloidose), kan oppstå ved kronisk betennelse, for eksempel ved revmatiske sykdommer. En annen type, kalt AL-amyloidose (primær amyloidose), oppstår uten annen underliggende sykdom og kan ha symptomer som kan forveksles med revmatisk sykdom. tillegg finnes det en sjelden form kalt lokalisert amyloidose, hvor amyloid kun produseres og avleires i en isolert del av kroppen. Denne formen har generelt god prognose. Amyloidose utredes og følges opp hos spesialist innen det/de mest angrepne organer, ev. også hos hematolog. Revmatologen bør kunne gjenkjenne symptomene og skille dem fra revmatiske bindevevssykdommer, vaskulitt og artritt-sykdommer.
Forekomst
En svensk undersøkelse fant ant antall nye tilfeller var 1-2 per 100 000 innbyggere. AL-amyloidose er vanligere enn AA type. Høyeste forekomst blant 60-80 åringer (Hemminki K. 2012).
Amyloidose kan ses ved kronisk betennelse ved revmatiske sykdommer (type serum amyloid A (SAA)/ AA-amyloidose/ sekundær type). Den primære formen (systemic light-chain, AL-type) har sykdomstrekk som kan mistolkes som revmatisk sykdom (vennligst se differensialdiagnoser nedenfor). AA-amyloidose er relatert til mangeårig systemisk inflammasjon ved familiær middelhavsfeber, juvenil artritt, revmatoid artritt, Bekhterevs, Muckle Wells syndrom, inflammatorisk tarmsykdom og lignende. Lokalisert amyloidose er sjelden og defineres ved at amyloid er produsert og deponert i en isolert del av kroppen. I blodet foreligger ikke monoklonale, lette kjeder og prognosen er utmerket.
Symptomer (både AA og AL-amyloidose)
Både AA- og AL-amyloidose kan gi en rekke symptomer, avhengig av hvilke organer som er rammet.
- Hjerte: Hjertesvikt, angina, lavt blodtrykk, hjerterytmeforstyrrelser (referanse; Deshai HV, 2010).
- Nyrer: Tap av proteiner i urinen (proteinuri), som kan føre til hevelser, spesielt i ankler og legger. Nefrotisk syndrom med smertefrie hevelser/ødemer, særlig i ankler og legger (referanse: Fedotov SA, 2022).
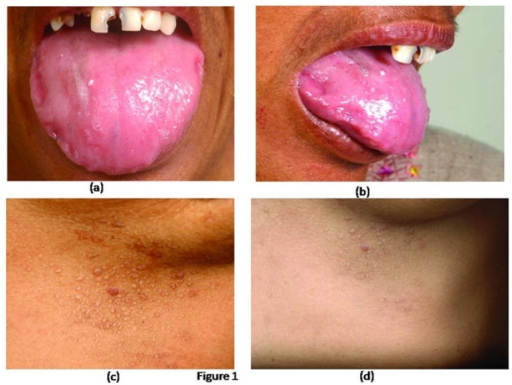
- Lever, milt, tunge, spyttkjertler: Forstørrelse av organene (hepatomegali, splenomegali, makroglossi) og redusert funksjon (munntørrhet) på grunn av amyloidavleiring.
- Nervesystem: Nummenhet og nervesmerter i hender og føtter (perifer nevropati, sensorisk polynevropati), vekttap (ved tarmplager), utmattelse, svimmelhet ved oppreising (autonome nervesystem), impotens (referanse: Al Hamed R, 2021). Carpal tunnel syndrom (nummenhet og smerter i hender)
- Skuldre: Amyloide skulderputer, skulderpute symptom med rotatorcuff fortykkelse kan påvises ved ultralyd eller MR og skyldes også amyloid-avleiringer (referanse: Liepnieks JJ, 2008).
- Hender: Voksaktig hud. Fingre med bøye-kontrakturer (palmarfascie), knuter på strekkesiden av underarmene.
- Hud: Blødninger (purpura, ekkymoser), små knuter under huden (papler), pigmentforandringer. Andre symptomer er voksaktig fortykkede fingre.
- Muskler. Forstørret muskelvolum, men mindre kraft (pseudohypertrofi).
- Skjelett: Bencyster som kan føre til benbrudd.
- Slimhinner: Tørrhet (øyne, munn), blødninger, forstørret tunge (makroglossi), redusert næringsopptak, vekttap.
- Lunger: Hoste, tung pust.
- Skjoldbruskkjertel og binyrer: Forstørrelse og funksjonsforstyrrelser.
{kind=link}
Undersøkelser
Diagnostisering av amyloidose innebærer en kombinasjon av sykehistorie, kliniske undersøkelser og ulike tester.
- Sykehistorien: registrering av aktuelle symptomer.
- Kliniske undersøkelser inkluderer systematisk vurdering av munn og tunge, hud, skuldre, hender, hjerte, lever og milt.
- Blodprøver: Celletellinger, lever- og nyrefunksjonsprøver, albumin, samt NTPro-BNP og troponin T, proteinelektroforese.
- Urinprøver: undersøkes for proteinutskillelse og protein/kreatinin clearance, urinelektroforese.
- EKG: Kan avsløre rytmeforstyrrelser ved hjertepåvirkning.
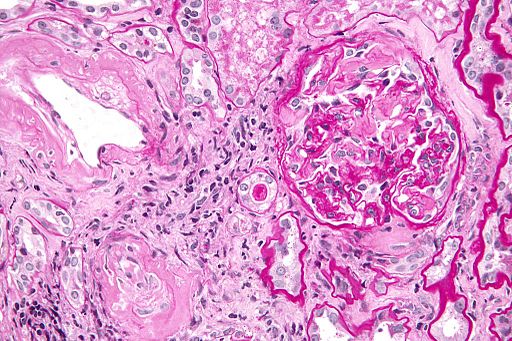
- Vevsprøver (biopsi). Diagnosen bekreftes vanligvis ved biopsi, ofte fra fettvev, endetarm eller spyttkjertler (referanse: Hallouch, RA). Biopsitakningen er vanligvis ukomplisert, men en bør være oppmerksom på generell økt blødningsrisiko ved amyloidose Prøvene blir farget med Congo rødt og undersøkt under mikroskop med polarisert lys, men også elektronmikroskopi og scintigrafi brukes.
- Andre undersøkelser: Avhengig av symptomer og organpåvirkning kan det være aktuelt med ytterligere undersøkelser, som for eksempel ultralyd, MR eller scintigrafi. Laser capture microdissection og tandem massespektrometri (LCM-MS) brukes for subtyping og er mer spesifikk enn anti-stoff baserte metoder/immunhistokjemi (referanse: Wisniowski B, 2020).
Amyloidose undergrupper
- Amyloidose kan deles i mer enn 30 typer avhengig av proteinet som danner amyloid. De vanligste er:
- Amyloid A (AA)- type: Forårsaket av annen, kronisk sykdom (sekundær amyloidose). Vennligst se under «Definisjon» overfor.
- Amyloidose (AL)-type): Forekommer uten annen sykdom (primær amyloidose). Mistenkes når serum (blodprøve) eller urin elektroforese viser monoklonal komponent av lette kjeder (λ, κ >95%) (Myelom. Mb. Waldenstrøm) og proteinuri eller andre suspekte symptomer og undersøkelsesfunn påvises (se over). Amyloid-vevet består av lett kjeder. Nefrotisk syndrom med mye protein utskillelse i urin påvises ofte. Benmargsbiopsi gjøres ofte som del av utredningen.
Andre amyloidose-relatere tilstander
- Familiær amyloidose (arvelig type)
- Beta-2-mikroglobulin-relatert amyloidose (ved langvarig dialyse)
- Aldersrelatert (senil) amyloidose (ofte med hjertesvikt)
- Alzheimer sykdom med amyloid avleiring i blodårer i hjernen
- Lokalisert Hud-amyloidose
- Lokalisert hjerte-amyloidose
- Øye-amyloidose
- Urinblære-amyloidose (blødninger/blod i urinen)
- Organspesifikk amyloidose (andre organer)
{kind=link}
{kind=link}
Lignende sykdommer (differensialdiagnoser)
- Diabetes mellitus: En metabolsk sykdom karakterisert av høyt blodsukker over tid, som kan føre til skade på flere organer, inkludert hjerte, nyrer, nerver og øyne, og dermed gi symptomer som kan forveksles med amyloidose
- Glomerulonefritt (nyrebetennelse): En betennelsestilstand i nyrenes glomeruli (filternøster) som kan føre til proteinuri (proteiner i urinen), hevelser og nedsatt nyrefunksjon, noe som kan ligne på nyrepåvirkning ved amyloidose.
- Hypotyreose (lavt stoffskifte, myksødem): En tilstand med nedsatt produksjon av thyreoideahormoner, som kan gi symptomer som tretthet, vektøkning, hevelser (myksødem) og i sjeldne tilfeller forstørret tunge (makroglossi), som kan overlappe med noen manifestasjoner av amyloidose.
- Forstørret tunge (macroglossi) av annen årsak (genetisk, lavt stoffskifte/hypotyreose med flere): En unormal forstørrelse av tungen som kan skyldes en rekke årsaker, inkludert genetiske forhold, hypotyreose eller andre tilstander, og som er et av symptomene som også kan forekomme ved amyloidose.
- Restriktiv kardiomyopati (en form for hjertesvikt): En hjertesykdom der hjertemuskelen blir stiv og mindre elastisk, noe som hindrer hjertet i å fylles ordentlig med blod, og dermed gir symptomer som ligner på hjerteamyloidose, som for eksempel tungpustethet og redusert fysisk yteevne.
- Systemisk sklerose (sklerodermi): En autoimmun bindevevssykdom som fører til fortykkelse og herding av huden og indre organer, og som i noen tilfeller kan gi symptomer som ligner på amyloidose, spesielt når det gjelder påvirkning av hjerte, lunger og mage-tarmkanalen.
- Sklerødem: En hudsykdom karakterisert av fortykket og stram hud, ofte lokalisert til nakke, skuldre og overkropp, som kan ligne på noen hudmanifestasjoner som kan forekomme ved amyloidose.
- Sjøgrens syndrom: En autoimmun sykdom som primært angriper de eksokrine kjertlene som produserer tårer og spytt, men som også kan påvirke andre organer, og i sjeldne tilfeller gi symptomer som ligner på amyloidose, for eksempel nevropati eller nyrepåvirkning.
Behandling
Behandlingen av amyloidose avhenger av den underliggende årsaken, typen amyloidose og pasientens tilstand.
AA-amyloidose: Behandlingen retter seg mot å kontrollere grunnsykdommen for å redusere risikoen for forverring og amyloidavleiring. Dette kan innebære bruk av immundempende legemidler, for eksempel IL-6 hemmer (tocilizumab) (referanser: Inoue D, 2010; Redondo-Pachón MD, 2013). Ved Familiær Middelhavsfeber (FMF) kan kolkisin være effektivt. Ved kronisk, inflammatorisk autoimmun sykdom er immundempende behandling viktig. Metotreksat og biologiske medikamenter som TNF-hemmere, rituksimab og spesielt tocilizumab (RoActemra) har vist seg effektive ved å kunne fjerne serum amyloid-A og kan vurderes som mer spesifikk terapi mot amyloiddepotet.
AL amyloidose: Behandlingen kan omfatte autolog stamcelletransplantasjon for enkelte pasienter, eller andre medikamentelle behandlinger. Dette innebærer at alderen ikke er for høy og at hjerte, nyrer og lunger fungerer godt nok. Samlet sett tilbys ca. 20% slik behandling (referanse: Al Hamed R, 2021; Ihne S, 2020).
Oppfølging og prognose
Pasienter med amyloidose trenger regelmessig oppfølging med blant annet scintigrafi, blod- og urinprøver for å overvåke sykdomsaktivitet og organfunksjon.
Prognosen ved amyloidose varierer avhengig av type, organpåvirkning og sykdomsutbredelse. Systemisk amyloidose er generelt en alvorlig tilstand, men tidlig diagnose og behandling kan forbedre utsiktene.
Litteratur
- Holt MF, 2023 (hjerte-amyloidose)
- Bustamante JG, 2020
- Ihne S, 2020
- Wechalekar AD, 2016
- Real de Asúa, D, 2014
- Merlini G, 2011
- Husby G, Tidsskriftet 1996
- Grans Kompendium i Revmatologi