

Definisjon
Interstitiell pneumoni med autoimmune kjennetegn/features (IPAF) er en sykdomsgruppe der det foreligger en kombinasjonen av idiopatisk interstitiell pneumoni (IIP) og en bakenforliggende ikke nærmere klassifisert systemisk bindevevssykdom (referanse: Fisher A, 2015). Tilstanden er et eksempel på at samarbeid mellom lungelege og revmatolog er nyttig i utredning og diagnostisering.
Bakgrunn
I 2015 bestemte European Respiratory Society/American Thoracic Society at interstitial pneumonia with autoimmune features (IPAF) skulle beskrive interstitiell lungesykdom ILD kombinert med ikke nærmere definert autoimmun sykdom. Det innebærer at en slik bakenforliggende sykdom ikke er klart definert. Dermed passer utslagene i blodprøver, bildediagnostisk og/eller vevsprøver ikke klart inn i en spesifikk bindevevssykdom selv om noen kjennetegn er til stede (referanse: Fischer A, 2015).
Symptomer
Sykdomstegn fra lunger varierer fra litt økt pustebesvær og/eller tørrhoste ved fysisk tung belastning til alvorlig lungesvikt. Revmatiske symptomer kan være ledd– eller muskelsmerter, Raynauds fenomen, utslett, feber eller symptomer fra ulike organer.
Undersøkelser
Sykehistorien omfatter aktuelle symptomer (se ovenfor) fra lunger og fra ledd, muskler, hud og andre organer.
Klinisk undersøkelse kartlegger eventuelle forandringer i hendene som sprekker og flassende hud på fingre i form av fissurer og “mekaniker hender” (antisyntetase syndrom), sår / nekroser (systemisk sklerose), leddbetennelser (artritt), Raynauds fenomen (systemisk sklerose, MCTD), blodåretegninger (teleangiektasier ved systemisk sklerose, begrenset form), hovne/puffy fingre (MCTD, systemisk sklerose) og rød-fiolette fargeforandringer (livide) hudforandringer over fingrenes grunn- og mellomledd (hhv MCP og PIP-ledd) (Gottrons tegn ved dermatomyositt).
Laboratorieprøver kan omfatte celletellinger (røde og hvite blodlegemer, blodplater), lever- nyre- og thyreoidea (stoffskifte)-funksjonsprøver, CRP, SR og kreatin kinase (CK), samt urin stiks (proteiner eller blod). Antistoff-tester omfatter ANA som ved positiv test undersøkes for ENA og subgrupper. Myositt-spesifikke antistoff og/eller systemisk sklerose-blot bør også vurderes. Ved artritt undersøkes revmafaktorer (RF) og anti-CCP antistoff. Ved mistanke om vaskulitt i små eller mellomstore kar suppleres med ANCA (PR3 og MPO) (referanse: Jee AS, 2017).
Lungefunksjonstester via lungelege vil tyde på nedsatt lungefunksjon som bør nærmere utredes med bildediagnostikk (se nedenfor).
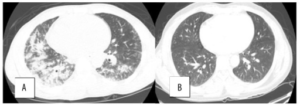
Bildediagnostikk er viktig. CT av lunger ofte i form av high-resolution chest tomography (HRCT) vil oftest vise tegn til non-spesifikk interstitiell pneumoni (NSIP), organisert pneumoni (OP) eller lymfocytisk interstitiell pneumoni (LIP) (referanse: Sambataro G, 2019). Pleura– og perikardvæske kan også være en del av tilstanden.
Diagnosen IPAF
Diagnosen stilles på grunnlag av sykehistorie, klinisk undersøkelse, laboratorieprøver og bildediagnostikk.
Det er vanligvis enighet om at følgende tre punkter skal oppfylles:
- Idiopatic Interstital Pneumonia (IIP) skal være diagnostisert på bakgrunn av
- HRCT undersøkelser av lunger og/eller NSIP (non-spesifikk interstitiell pneumoni) -lignende bilde er vanligst, men UIP («usual interstitial pneumonia») er heller ikke uvanlig i sykdomsforløpet.
- Vevsprøve (biopsi) fra lungevev
- Systemisk, ikke klassifiserbar bindevevssykdom diagnostiseres på bakgrunn av minst to av tre (A-C) og punkt 3. nedenfor:
- A. Symptomer på revmatisk sykdom
- B. Antistoff (oftest ANA, SSA/Ro52)
- C. Sykdomstegn ved undersøkelse (for eksempel Raynauds fenomen, artritt, eksem/dermatitt og sykdomstegn ved kapillaroskopi)
- Andre sykdomsårsaker må utelukkes ved grundig klinisk vurdering
- Klassifikasjonskriterier for andre systemiske bindevevssykdommer (systemisk sklerose, myositt, antisyntetase syndrom, Sjøgrens sykdom, revmatoid artritt og andre) skal ikke oppfylles
- Infeksjoner
- Bivirkning av medikamenter
I forskning brukes klassifikasjonskriterier. Disse kan brukes med forsiktighet også ved diagnostisering i klinisk hverdag når diagnosen på forhånd mistenkes (se nedenfor):
Foreslåtte klassifikasjonskriterier for IPAF (Fisher A, 2015).
For å oppfylle kriterier for IPAF kreves:
- Radiologisk eller histopatologisk tegn på interstitiell pneumoni og
- Komplett klinisk vurdering som utelukker andre årsaker til interstitiell pneumoni og
- Inklomplette tegn til typisk systemisk bindevevssykdom (utelukke sikker SLE, Sjøgrens, (dermato-) myositt, systemisk sklerose, MCTD)
I tillegg til alle tre punkter ovenfor kreves minst ett punkt fra minst to av de tre domener i kolonne A, B og C i tabellen nedenfor:
| A. Kliniske kjennetegn | B. Serologi | C. Morfologiske manifestasjoner |
|---|---|---|
| 1. Distale digitale fissurer (mekaniker-hender)2. Distale digitale ulcerasjoner på fingerpulpa3. Inflammatorisk artritt eller polyartikulær leddstivhet om morgenen >60 minutter4. Palmar telangiektasi5. Raynauds fenomen6. Uforklarte digitale ødemer / puffy hands7. Uforklart vedvarende utslett over fingrenes DIP-ledd (Gottrons tegn) | 1. ANA ≥1: 320 titer, diffus, speckled, homogenent mønster or a) ANA nukleært mønster (uansett titer) eller b) ANA centromer mønster (uansett titer) 2. Revmatoid factor ≥2× øvre nivå av referanseområdet 3. Anti-CCP 4. Anti-dsDNA 5. Anti-Ro (SS-A) 6. Anti-La (SS-B) 7. Anti-ribonucleoprotein (RNP) 8. Anti-Smith (Sm) 9. Anti-topoisomerase (Scl-70) 10. Anti-tRNA syntetase ( Jo-1, PL-7, PL-12, EJ, OJ, KS, Zo, tRS) 11. Anti-PM-Scl 12. Anti-MDA-5 | 1. Typisk radiologisk mønster ved HRCT a) NSIP b) OP c) NSIP med OP overlapp d) LIP 2. Histopatologisk mønster eller funn ved kirurgisk lungebiopsi: a) NSIP b) OP c) NSIP med OP overlapp d) LIP e) Interstitielle lymfoide aggregater med germinale sentre f) Diffus lymfoplasmacytisk infiltrasjon (med- eller uten lymphoide follikler) 3. Multi-compartment manifestasjon (i tillegg til interstitiell pneumoni): a) Uforklart pleuravæske eller pleurafortykkelse b) Uforklart perikardvæske eller perikardfortykkelse c) Uforklart interstitiell lungesykdom d) Uforklart pulmonal vaskulopati |
LIP: lymfoid interstitiell pneumonia NSIP: non-spesifikk interstitiell pneumoni. OP, organiserende pneumoni.
Lignende tilstander / differensialdiagnoser ved IPAF
Symptomer og funn ved IPAF overlapper med andre tilstander. En bør skille IPAF fra følgende tilstander:
- Idiopatisk pulmonal fibrose (IPF)
- Lungesykdom som er relatert til spesifikke systemisk bindevevssykdommer (CTD-ILD) som antisyntetase syndrom, dermatomyositt og systemisk sklerose
- Cryptogen organisert pneumoni (COP)
- Idiopatisk non-spesifikk interstitiell pneumoni (NSIP)
- Lymfocytisk interstitiell pneumoni (LIP) (Ofte ved Sjøgrens syndrom)
Behandling av IPAF
Lungeforandringene ved IPAF strekker seg fra inflammasjon (revmatisk betennelse) til fibrose med økt bindevev. Mens inflammasjon lar seg behandle med kortikosteroider (Prednisolon) og andre immundempende medikamenter, er fibrose mindre påvirkelig. Det finnes imidlertid anti-fibrotiske medikamenter som kan redusere eller stanse utviklingen (referanse: Shehata M, 2021). Gode studier foreligger ikke ennå, og behandlingen er således utprøvende (Referanse: Torrisi SE, 2019).
Det anbefales å vurdere behandlingsstart dersom lungeforandringene øker og omfatter 20% eller mer av lungevevet (ved HRCT). Valg av medikament baseres på vurdering av den enkeltes sykdomsforløp og gjøres av erfarne leger, oftest spesialister i lungesykdommer i samråd med spesialister i revmatologi. Medikamenter som ofte vurderes er: Kortikosteroider (Solu-Medrol, Prednisolon), Cyclofosfamid, Rituximab, Mykofenolat, Azathioprin, Ciclosporin A.
Ved alvorlig forløp vil noen trenge lungetransplantasjon (referanse: Atienza-Matheo B, 2020).
Oppfølging
Dersom lungeforandringene er de mest alvorlige manifestasjonen, er regelmessig oppfølging hos lungelege essensielt. HRCT undersøkelser og lungefunksjonsundersøkelser, eventuelt supplert med 6-minutters gangtest benyttes ofte. Revmatologer bidrar ofte med lang erfaring med de immundempende medikamentene og vurdering av ulike sykdomsmanifestasjoner utenom lungene.
Det foreligger begrensede data, men foreløpige resultater tyder på at overlevelsen ved IPAF er sammenlignbar med systemisk bindevevssykdommer med tilsvarende lunge-manifestasjoner og bedre enn ved idiopatisk lungefibrose (Wells AU, 2020).
Litteratur
- Shehata M, 2021
- Fisher A, 2015
- Collins B, 2016
- S-M Koo, 2017
- Fischer A, 2013
- Grans Kompendium i Revmatologi