


Definisjon
Histiocytose er en gruppe sjeldne sykdommer der histiocytter som er spesielle hvite blodlegemer ved en feil lagres i store mengder i ulike organer. Histiocytose ble tidligere (WHO 1987) delt inn i tre hovedgrupper: 1. Langerhans histiocytose. 2. Non-Langerhans. 3. Malign (ondartet) histiocytose. Senere er det foreslått en inndeling i fem hovedgrupper (referanse: Emile J-F, 2016). Tidligere navn på histiocytose-sykdommene er Histiocytose-X, Letter-Siwe sykdom, Hand-Chüller-Christian sykdom og diffus retikulo-endoteliose. Tilstandene utredes ofte a spesialist i blodsykdommer, men revmatologer forespørres ved revmatiske symptomer. Sykdommen kan være vanskelig å diagnostisere.
Forekomst
Histiocytose defineres som en sjelden sykdom. Langerhans histiocytose er den vanligste formen med forekomst (insidens) på 1-2 nye tilfeller pr. million personer årlig (referanse: Tillotson CV, 2021).
Symptomer
Symptomer og undersøkelsesfunn varierer avhengig av hvilken type som foreligger.
- Vanligste sykdomsdebut er i barne- og tidlig ungdomsalder.
- Enkelte typer kan debutere blant voksne slik som Erdheim-Chesters sykdom.
- Pulmonal histiocytose (undergruppe av Langerhans cellehistiocytose, angriper lunger).
- Retikulo-histiocytose kan ligne leddgikt, RA).
Undersøkelser
Sykehistorien omfatter gradvis debut og typiske symptomer (se ovenfor).
Klinisk gjøres en generell undersøkelse med særlig vurdering av hud, lymfeknuter, skjelett med skalle og indre organer med lever og milt.
Blodprøver viser ofte tegn på betennelse med forhøyet CRP og SR, men ikke utsalg i «revma-prøver» / antistoff eller andre spesielle tester.
Bildediagnostikk omfatter CT og MR-undersøkelser av hode, hals, bryst- og mage-områder, avhengig av mistenkte manifestasjoner. Også PET/CT kan være nyttig for å kartlegge sykdomsutbredelsen.
Vevsprøver (biopsi) er oftest avgjørende for diagnosen. Prøven kan tas fra angrepet område i hud, lymfeknute eller benmarg. Vevsprøver (biopsi) beskrives ved histiocytose i blant som «eosinofilt granulom».
Diagnosen
Diagnosen stilles på bakgrunn av symptomer og undersøkelsesfunn (se ovenfor)
1.»L» (Langerhans) gruppen
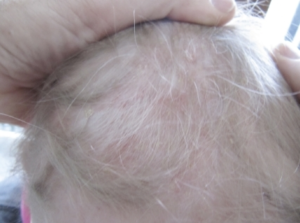
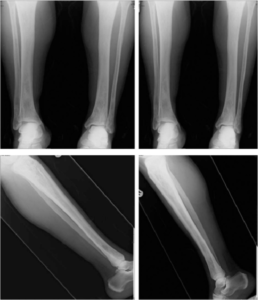
Symptomer, undersøkelsesfunn og alvorlighetsgrad varier fra harmløst til alvorlig. Utslett er ofte debutsymptom, feber, vekttap, mange skjeletthevelser (ca. 80%), oftest i hodet/skallen, men også i hofter/bekken, lårben eller ribben. Eksem og sår. Stor lever og milt (hepatosplenomegali) og lymfeknuteforstørrelser. Lunger der bildediagnostikk kan vise knuter/nodulære fortetninger (pulmonal histiocytose X). Mer enn halvparten av tilfellene oppstår blant barn før 10 års alder.
Diagnosen bekreftes ved vevsprøve (biopsi), oftest fra hud. Vevet farges med S-100 og CD-1a. Elektronmikroskopisk ses såkalte cytoplasmatiske Birbeck granula.
Lignende tilstander / differensialdiagnoser er acrodermatitis enteropathica, acropustulosis blant nyfødte, medfødt candidiasis, eosinofil pustuløs follikulitt, incontinentia pigmenti, leukemi, lymfom, mastocytose, myelom og neonatal pustulær melanose.
Behandlingen varierer mellom observasjon, medikamenter, kirurgi og strålebehandling, avhengig av alvorlighetsgrad.
Litteratur: Tillotson CV, 2022.
Dette er en multiorgansykdom som angriper menn hyppigere enn kvinner Debutalder er i gjennomsnitt 55 år (16-80 år). Typisk er skjelett-sklerose i de lange rørknokler og hevelse omkring nyrer («hairy kidneys»). Vennligst les mer på egen side om Erdheim-Cehsters sykdom her
2. «C» gruppen (Cutan (hud) og mucocutan (hud og slimhinner)
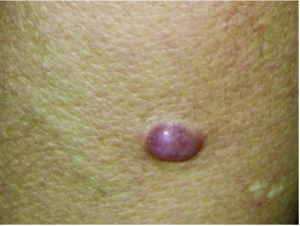
Retikulo-histiocytose, begrenset- og multisentrisk type)
Multisentrisk type: Alvorlig artritt (leddbetennelser) i hender. Utbredte hud- og betydelige artritt-forandringer. Vanligvis kvinner 50-60 år. Varierende, svingende sykdomsaktivitet over flere år. Retikulo-histiocytose (begrenset og multisentrisk type); vennligst les mer i eget kapittel.
Xantogranulom gruppen: Ulike former som kan angripe fra små barn til voksne. Enkelte eller mange røde-gule knuter i huden. 0,5-1,0 cm i diameter. Dersom også andre organer omfattes, innordnes sykdommen i «L» Gruppen (se ovenfor).
Sea-blue histiocytose; Hudaffeksjon med histiocytter i fettvev. Arvelig eller etter langvarig parenteral fettrik ernæring.
3. «M» (Malign/kreft) Gruppen. Malign histiocytose
Sekundære former ved lymfom og leukemi. Subtype av akutt myeloid leukemi. Symptomer omfatter hoste, redusert appetitt, slapphet og tung pust. Klinisk påvises forandringer i lunger, lymfeknuter, lever, milt og nervesystem. Undersøkelser kan vise sykdomstegn i lunger, lymfeknuter, lever, milt og hjernen. Vevsprøve viser histiocytt-infiltrasjon. En skiller primære former sekundære former ved lymfom.
4. «R» (Rosai-Dorfman) gruppe og forskjellige former som ikke angriper hud
Rosai Dorfman Sykdom, Sinus histiocytose med massiv lymfadenopati (SHML). Definisjon: Sjelden sykdom med mange ulike manifestasjoner der blant annet lymfeknuter hovner opp. Vevsprøve (biopsi) som er avgjørende for diagnosen viser et typisk bilde med histiocytter som inneholder lymfocytter. Angriper vanligst barn og unge voksne. Symptomer omfatter fFeber, nattesvette, tretthet og vekttap. Undersøkelsesfunn kan vise varierende forandringer som hovne lymfeknuter, oftest på halsen. Hudforandringer med lokalisert tap av fett, tetthet i nese og bihuler, trykk bak øyne og dobbeltsyn, forstørret milt og osteonekrose. Diagnosen baseres på sSymptomer og undersøkelsesfunn Vevsprøve (biopsi) er avgjørende. Lignende tilstander / differensialdiagnoser er GPA (Wegeners granulomatose), Pannikulitt, Langerhans histiocytose (se ovenfor), lymfom, sarkoidose, tuberkulose og IgG4 relatert sykdom. Behandling er med immundempende medikamenter. Kortikosteroider kan være tilstrekkelig hos noen.
5. «H» Gruppen. Hemofagocytisk lymfohistiocytose (HLH) og Makrofag aktiverings syndrom (MAS)
Cytokin storm med ukontrollert aktivering av lymfocytter og makrofager (spesielle typer hvite blodlegemer i immunsystemet). Se også makrofag aktiverings-syndrom (MAS).
Annet
Histiocytter ses også ved Kikutchi-Fujimoto sykdom (KFS) er en sjelden, benign, nekrotiserende lymfadenopati (sykdom i lymfeknuter), oftest på halsen hos unge kvinner. Histologi (vevsundersøkelse ved biopsi) viser histiocytter og nekrotiserende non-maligne funn.
Litteratur
- Emile J-F, 2016
- Allen CE, 2015 (Behandling)
- Grischikowsky M, 2015 (Management)
- Grans Kompendium i Revmatologi