


Definisjon
Periodiske febersyndromer, også kalt autoinflammatoriske sykdommer, er en gruppe sjeldne tilstander som kjennetegnes av gjentatte episoder med feber uten at man finner tegn til infeksjon. Typisk forløp er feberepisoder med ganske like lange feberfrie perioder imellom. I tillegg til feber kan man oppleve symptomer som halsbetennelse, munnsår, mageproblemer, ledd- og muskelsmerter, hodepine, hudutslett, hovne lymfeknuter og utmattelse. Betennelsesprøvene (CRP og SR) er forhøyet under anfallene. Sykdommene debuterer vanligvis i barne- og ungdomsårene, men kan i noen tilfeller oppstå først i voksen alder. For å stille riktig diagnose er det viktig å vurdere sykehistorie, symptomer, alder ved debut og resultater fra genetiske analyser. Periodiske febersyndromer skiller seg fra autoimmune (systemiske) bindevevssykdommer og vaskulitt, men fellesnevneren er at immunsystemet angriper kroppens eget vev, noe som fører til betennelse. Heldigvis finnes det effektiv behandling mot flere av disse tilstandene.
Sykdomsårsak
De fleste periodiske febersyndromer skyldes en mutasjon (endring) i ett enkelt gen (arvestoff). Unntakene er PFAPA og Stills sykdom/systemisk JIA, hvor flere gener er involvert. Mutasjonen fører til at det medfødte (innate) immunsystemet mangler viktige bremser, og dermed reagerer uten at kroppen har vært i kontakt med bakterier, virus eller andre fremmede stoffer.
Forekomst
Periodiske febersyndromer er sjeldne sykdommer. Familiær Middelhavsfeber (FMF) er den vanligste monogene (skyldes ett gen) autoinflammatoriske sykdommen på verdensbasis (Mortensen SB, 2020). Hos små barn er «periodisk feber, aftøs stomatitt, faryngitt og adenitt syndrom» (PFAPA) det vanligste periodiske febersyndromet (referanse: Rigante D, 2009).
Symptomer
Symptomene på periodiske febersyndromer varierer, men noen fellestrekk er:
Brå sykdomsdebut: Symptomene kommer ofte plutselig.
Episoder med feber og betennelse: Høy feber som kommer og går, utmattelse, halsbetennelse, hudutslett og leddbetennelse (artritt).
Ulike typer periodiske febersyndromer
De ulike periodiske febersyndromene kan skilles fra hverandre basert på hvordan feberen forløper, blodprøvefunn og hvilke organer som er involvert.
Febermønster:
- PFAPA: Ganske regelmessige feberanfall (hver 2-4 uke) som varer i 4-6 dager. Feberen og de andre symptomene blir ofte mildere med alderen.
- Familiær Middelhavsfeber (FMF): Kortest feberperioder (2-3 dager) med uregelmessige intervaller.
- TRAPS: Lengst feberperioder (opptil tre uker ved alvorlig form, 5-9 dager ved mild form).
- HIDS (Hyper IgD syndrom): Svært regelmessig feber med brå temperaturstigning.
Blodprøver
- Senkningsreaksjon (SR) og CRP er forhøyet under feberanfallene.
- Lett økning i antall hvite blodceller (leukocytose) kan forekomme.
- Det påvises ingen tegn til infeksjon.
Organ-symptomer
- Leddbetennelser (Artritt): Vanlig ved PAPA og Blau syndrom.
- Lungesykdom: Artritt, lungesykdom og lungeblødning: COPA (genetisk vaskulitt blant barn) (referanse: Vece TJ, 2016).
- Slimhinnesår: kan ligne Behcets sykdom (sår i slimhinner): kan sees ved A20 protein haploinsuffisiens (Referanse Zhou Q, 2015)
- Makrofag aktiveringssyndrom (MAS): Forekommer i noen tilfeller, spesielt ved NLRC4 mutasjoner.
- Betennelse i fettveva (pannikulitt): CANDLE
- Hudsykdommer: Pustuløs psoriasis: DIRA, DITRA
- Urticariell vaskulitt: Pustuløs psoriasis (DIRA, DITRA) og urticariell vaskulitt (FCAS, Muckle-Wells, CINCA).
- Vaskulitt og lungesykdom: Kan forekomme ved SAVI (STING-assosiert vaskulopati med begynnelse i barndom) (Wang Y, 2021)
Undersøkelser
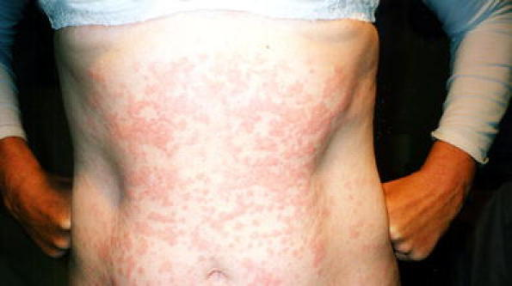
Utredning av periodiske febersyndromer gjøres ofte av barneleger i samarbeid med barnerevmatolog, øyelege, hudlege og genetiker. Undersøkelsene kan omfatte:
- Sykehistorie: Kartlegging av symptomer, alder ved debut, febermønster og familiehistorie.
- Klinisk undersøkelse: Generell undersøkelse for å utelukke infeksjoner og vurdere organmanifestasjoner.
- Blodprøver: Måling av betennelsesmarkører (CRP og SR), samt genetisk testing. Aktuelle tester er oppfør her (Genetikkportalen.no).
- Bildediagnostikk: Røntgen, ultralyd og CT kan visualisere betennelse i ulike organer.
- Benmargsundersøkelse: Kan være aktuelt ved usikker diagnose.
Behandling
Behandlingen avhenger av hvilket periodisk febersyndrom man har. Mulige behandlingsalternativer:
- Kortikosteroider (prednisolon): Prednisolon kan brukes til å dempe betennelsen.
- Biologiske legemidler: Anakinra (Kineret) og canakinumab (IL-1 hemmere) eller TNF-hemmere kan være effektive.
Ulike febersyndromer/autoimmune sykdommer
A20 haploinsuffisiens (HA20), TNFAIP3 mutasjon
Definisjon: A20 haploinsuffisiens (HA20) er en sjelden sykdom som fører til betennelser i kroppen. Den ble først beskrevet i 2016 og rammer oftest barn. Sykdommen skyldes en feil i et gen som heter TNFAIP3. Dette genet er ansvarlig for å lage et protein (NF-κΒ regulerende protein A20) som regulerer betennelsesreaksjoner i kroppen. Når genet ikke fungerer som det skal, oppstår det overdreven betennelse (referanse: Zhou Q, 2016). Debutalder: Vanligvis barn før 10 års alder, men kan debutere fra spedbarnsalder til unge voksne.
Symptomer:
- Sår: Residiverende (kommer ofte tilbake) smertefulle sår i munnen er vanligst. Sår i svelg og mage-tarm kan også forekomme.
- Magesmerter: Betennelse i mage-tarm kan gi magesmerter.
- Leddplager: Betennelse i ledd (artritt) kan gi smerter og stivhet.
- Hudsymptomer: Ulike hudutslett som pustler (små kviser med puss), follikulitt (betennelse i hårsekkene), akne og sår.
- Øyebetennelse: Betennelse i øyets regnbuehinne (iridocyklitt) og netthinne (retinal vaskulitt).
Hvordan stilles diagnosen? Diagnosen mistenkes ved typiske symptomer og funn ved legeundersøkelse. For å bekrefte diagnosen er det nødvendig med en genetisk test som kan påvise mutasjon i TNFAIP3 genet.
Andre sykdommer som kan ligne (differensialdiagnoser):
- Behcets sykdom
- Juvenil lupus (JSLE)
- Juvenil idiopatisk artritt (JIA)
- PFAPA syndrom
- Inflammatorisk tarmsykdom (IBD)
Behandling: Biologiske legemidler i form av IL-1 hemmere (anakinra, canakinumab) eller TNF-hemmere har effekt hos de fleste.
Litteratur: Zhou Q, 2016; Aeschlimann AF, 2018.
Behcets sykdom
Behcets sykdom regnes av noen som en autoinflammatorisk sykdom, spesielt den familiære formen tilhører de autoinflammatoriske sykdommene (referanse: Leccese P, 2019). Det gjelder spesielt den familiære formen A20 Haploinsuffisiens (HA20) som rammer barn. Dette til tross for at sykdommen oftest klassifiseres som en systemisk vaskulitt.
Blau syndrom (juvenil sarkoidose)
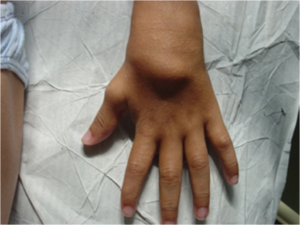
Blau syndrom er en sjelden genetisk sykdom som gir betennelse i kroppen. Den rammer oftest barn mellom 2 og 4 år. Sykdommen kjennetegnes av en triade av symptomer: betennelse i øynene (uveitt), leddbetennelse (artritt) og karakteristiske hudutslett. Når sykdommen oppstår sporadisk (uten kjent arv), kalles den neonatal sarkoidose eller pediatrisk granulomatøs artritt. Sykdomsårsak: Blau syndrom skyldes en mutasjon i et gen som kalles NOD2. Dette genet er viktig for immunforsvaret og bidrar til å regulere kroppens reaksjon på bakterier og andre fremmede stoffer. Debutalder: 2-4 år er vanligst.
Symptomer:
- Øyebetennelse: Betennelse i øyets regnbuehinne (uveitt) som ofte rammer begge øyne.
- Leddbetennelse: Betennelse i ledd (artritt) som kan gi smerter, hevelse og stivhet. Håndledd, fingerledd, ankler og albuer er ofte affisert. Sykdommen kan føre til redusert bevegelse i leddene (kontrakturer) tidlig i forløpet. I noen tilfeller kan det oppstå cyster i leddene som på sikt kan skade leddene (referanse: Wouters CH, 2014;
- Senebetennelse, spesielt over håndryggen og håndleddet, er også vanlig (referanse: Pac Kisaarslan PA, 2020).
- Hudutslett: Brunaktige, skjellende utslett og små knuter som kan oppstå over hele kroppen.
Diagnosen stilles basert på de typiske symptomene, bildediagnostikk og vevsprøver. Vevsprøver viser granulomer, som er en spesiell type betennelsesreaksjon. Genetisk testing kan bekrefte diagnosen ved å påvise mutasjoner i NOD2 genet.
Behandling med biologiske legemidler som hemmer betennelsesprosesser (IL-1 hemmere eller TNF-hemmere) er vanlig. I noen tilfeller brukes også legemidlet thalidomid.
Litteratur: Punzi L 2009; Wouters CL, 2014.
CANDLE, Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature
CANDLE syndrom er en sjelden og alvorlig genetisk sykdom som fører til kronisk betennelse i kroppen. Sykdommen starter vanligvis i løpet av barnets første leveår, ofte allerede i de første ukene. Sykdomsårsak: CANDLE syndrom skyldes en feil i et gen som heter PSMB8. Dette genet er ansvarlig for å lage en viktig del av cellenes «resirkuleringssystem». Cellene våre inneholder proteasomer, som er små «maskiner» som bryter ned proteiner som ikke lenger trengs eller er skadet. Når PSMB8-genet ikke fungerer som det skal, påvirkes denne prosessen, og det kan føre til opphopning av avfallsstoffer og betennelse. Debutalder: Starter i barnets første leveår, ofte de 2-4 første leveukene.
Symptomer
- Feber: Gjentatte feberepisoder, ofte daglig eller annenhver dag.
- Hudutslett: Røde, ringformede utslett som kan vare i dager eller uker.
- Hevelse: Hevelse rundt øynene, i fingre og tær, og forstørret lever.
- Nedsatt trivsel: Redusert vekst og utvikling.
- Fettvevssvinn: Tap av fettvev under huden (lipodystrofi).
- Hovne lymfeknuter: Forstørrede lymfeknuter.
Sjeldnere symptomer:
- Hevelse rundt munnen og i spyttkjertlene.
- Øyebetennelse.
- Mørkfarging av huden (acanthosis nigricans).
- Økt hårvekst.
- Leddsmerter.
- Betennelse i fettvev (pannikulitt).
- Flekkvis håravfall (alopecia areata).
Diagnosen stilles basert på de karakteristiske symptomene, kliniske funn og genetisk testing. Vevsprøver fra huden kan vise tegn på betennelse.
Behandling: Det finnes dessverre ingen effektiv behandling som kan kurere CANDLE syndrom. Behandlingen fokuserer på å lindre symptomene og forbedre livskvaliteten. Kortisonpreparater og metotreksat kan forsøkes. Noen pasienter kan ha effekt av biologiske legemidler som hemmer betennelsesstoffer (IL-1 hemmere, TNF-hemmere og IL-6 hemmere) (referanse; Liu Y, 2011).
Litteratur: Torrelo A, 2017.
CAPS
- Vennligst se CINCA nedenfor.
CINCA: Kronisk infantil nevrologisk kutant og artikulært syndrom / Neonatal-Onset Multisystem Inflammatory Disease (NOMID)
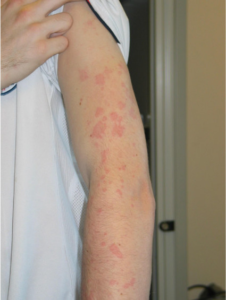
CINCA/NOMID syndrom er en sjelden og alvorlig genetisk sykdom som fører til kronisk betennelse i kroppen. Sykdommen tilhører en gruppe sykdommer som kalles kryopyrin-assosierte periodiske syndromer (CAPS). Muckle Wells syndrom (se nedenfor) er en mildere variant av CAPS. Sykdomsårsak: CINCA/NOMID skyldes vanligvis en mutasjon i et gen som heter NLRP3. Dette genet spiller en viktig rolle i reguleringen av betennelsesreaksjoner. Mutasjonen fører til overproduksjon av et signalstoff som kalles interleukin-1 beta (IL-1β), som forårsaker betennelse. I 30-40 % av tilfellene finner man imidlertid ikke NLRP3-mutasjon. Andre gener, som for eksempel NLRP12, kan også være involvert (vennligst se NLRP12 assosiert febersyndrom nedenfor). Forekomst (prevalens): 12-/million.
Symptomer: De fleste får symptomer allerede i løpet av de første dagene etter fødselen.
- Hudutslett: Kronisk elveblestlignende utslett.
- Feber: Mild feber.
- Leddsmerter: Smerter og stivhet i ledd.
- Muskelsmerter: Smerter og ømhet i muskler.
- Øyebetennelse: Betennelse i øyet (uveitt).
- Hjernehinnebetennelse: Kronisk betennelse i hjernehinnene (meningitt).
- Forsinket utvikling: Fysisk og/eller mental utviklingshemming.
- Karakteristisk ansiktsform: Noen utvikler en karakteristisk ansiktsform med fremtredende panne.
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing.
Behandling: Behandling med biologiske legemidler som blokkerer IL-1 (anakinra, canakinumab) har god effekt hos mange.
Litteratur: Finetti M, 2016.
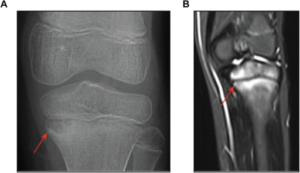
CRMO (chronic multifocal osteomyelitis)
CRMO (Kronisk residiverende multifokal osteomyelitt) er en sjelden sykdom som forårsaker betennelse i skjelettet. Sykdommen rammer oftest barn og unge, og kan lett forveksles med en infeksjon. Hos voksne klassifiseres tilstanden oftere som en undergruppe av SAPHO syndromet. Debutalder: Barn etter 2 års alder. De fleste er mellom 8 og 13 år. Median alder ved diagnose er 10 år. Forekomst: Sjelden sykdom, men økende oppmerksomhet og bedre diagnostikk (MR) medfører at flere tilfeller er diagnostisert i senere tid. Årsak: Den eksakte årsaken til CRMO er ukjent, men man tror at det skyldes en feil i immunforsvaret som fører til at kroppen angriper sitt eget vev. I noen tilfeller har man funnet en genetisk mutasjon som kan bidra til sykdommen. Denne mutasjonen fører til økt produksjon av betennelsesstoffer.
Symptomer:
- Smerter: Smerter i skjelettet eller ledd, ofte verst om natten eller morgenen.
- Hevelse: Hevelse over det betente området (artritt) hos ca. 30%.
- Nedsatt bevegelighet: Stivhet og redusert bevegelighet i ledd.
- Brudd: I noen tilfeller kan sykdommen debutere med et brudd uten noen klar årsak.
Diagnose: CRMO er en eksklusjonsdiagnose, det vil si at man må utelukke andre mulige årsaker til symptomene før man kan stille diagnosen. Bildediagnostikk: Røntgen, CT og MR brukes for å visualisere betennelsen i skjelettet. MR er den mest sensitive metoden og kan vise betennelse tidlig i forløpet. Blodprøver: Blodprøver kan vise tegn på betennelse, men er ofte normale. Vevsprøve: I noen tilfeller kan det være nødvendig å ta en vevsprøve (biopsi) for å utelukke andre sykdommer som infeksjon eller kreft. Genetikk: I noen tilfeller kan heterozygot mutasjon i det filamin-bindende domenet påFBLIM1 genet påvises.
Andre sykdommer som kan ligne (differensialdiagnoser): Leukemi, lymfom, primære og sekundære skjelettsvulster som Ewing sarkom, rhabdomyosarkom, neuroblastom og metastaser. Godartede svulster er eosinofilt granulom og osteoid osteom. Infeksjoner omfatter bakterie infeksjon i skjelettet (osteomyelitt) inklusiv tuberkulose (tbc). Langerhans histiocytose, PAPA, DIRA (se nedenfor) og Majeed syndrom (mutasjon i LIPIN2 genet) er sjeldne tilstander som også ligner CRMO. Også sirkulasjonsforstyrrelser i skjelettet med osteonekrose som ved Legg-Calve Perthes sykdom utelukkes. Økt forkalkning i skjelettet (osteosklerose) ved histiocytose, sigd-celle sykdom og osteopetrose kan ligne reparasjons-prosess ved CRMO.
Behandling: Betennelsesdempende medisiner: NSAIDs (ikke-steroide antiinflammatoriske legemidler) er ofte førstevalget i behandlingen. Kortison: Kortison kan brukes i perioder med mye betennelse. DMARDs: DMARDs (sykdomsmodifiserende antirevmatiske legemidler) som metotreksat eller sulfasalazin kan brukes hos noen. TNF-hemmere: Biologiske legemidler som hemmer TNF (tumor nekrosefaktor) kan være effektive hos noen pasienter. Bisfosfonater: Legemidler som styrker skjelettet kan brukes for å forebygge brudd.
Forløp og prognose: CRMO er en kronisk sykdom med et varierende forløp. Mange opplever perioder med bedring (remisjon) og tilbakefall. Prognosen på lang sikt er god, men noen kan utvikle komplikasjoner som skjelettdeformiteter, kroniske smerter og brudd i ryggsøylen.
Litteratur: Johnsson A, 2015; Hofmann SR, 2017; Koryllou A, 2021.
DADA-2, Autoinflammasjon med immunsvikt
Hjerneslag, tilbakevendende feber, marmorert hud, mild immunsvikt. Vennligst les om DADA2 på egen side.
DIRA (deficiency of the IL-1R antagonist)
DIRA (Deficiency of the IL-1 Receptor Antagonist) er en svært sjelden genetisk sykdom som fører til alvorlig ikke-infeksiøs betennelse (multifokal osteomyelitt med periostitt og pustulose) i kroppen. Sykdommen rammer nyfødte og viser seg vanligvis i løpet av de første ukene etter fødselen. Sykdomsårsak: Mutation i IL1RN genet som koder for interleukin-1 (IL-1) reseptor antagonist (IL-1Ra). IL-1Ra mangel medfører in høy inflammatorisk aktivitet via IL-1 og IL-1 reseptor (IL-1R1). Forekomst: Debut mellom de første dager etter fødsel og innen få uker.
Symptomer:
- Hudbetennelse: Eksem og pustler (små kviser med puss).
- Skjelettbetennelse: Betennelse i skjelettet (osteomyelitt) som kan ligne på en bakteriell infeksjon.
- Bindevevsbetennelse: Betennelse i bindevevet rundt knoklene (periostitt).
- Andre symptomer: I noen tilfeller kan det oppstå dannelse av nytt benvev (heterotrofisk bennydannelse), blodpropp (trombose) og betennelse i blodårene (vaskulitt) (referanse: Jesus AA, 2011).
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing. Genetisk testing kan påvise mutasjoner i IL1RN-genet.
Behandling: Behandling med biologiske legemidler som blokkerer IL-1 (anakinra, canakinumab) er svært effektivt. Hvis barnet ikke tåler disse medisinene, kan man forsøke hyposensibilisering, en metode som gradvis venner kroppen til medikamentet. Ubehandlet kan DIRA føre til alvorlige komplikasjoner som kronisk lungesykdom og blodpropp.
Litteratur: Mendonca LO, 2017.
DITRA (Deficiency of interleukin-36 receptor antagonist)
DITRA (Deficiency of Interleukin-36 Receptor Antagonist) er en sjelden genetisk sykdom som fører til betennelse i huden. Sykdommen rammer oftest gutter og debuterer vanligvis rundt 7 måneders alder. Gutter angripes oftere enn jenter. Årsak: DITRA skyldes en mutasjon i et gen som heter IL36RN. Dette genet er ansvarlig for å lage et protein som blokkerer effekten av interleukin-36 (IL-36), et signalstoff som er viktig for betennelsesreaksjoner i huden. Når IL36RN-genet ikke fungerer som det skal, fører det til overdreven IL-36-aktivitet og dermed hudbetennelse.
Symptomer:
- Feber: Episoder med feber.
- Pustuløst utslett: Utslett med små kviser fylt med puss (pustler) på håndflatene og fotsålene (palmoplantar pustulose).
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing. Genetisk testing kan påvise mutasjoner i IL36RN-genet.
Behandling: Kortison: Kortisonpreparater (steroider) kan dempe betennelsen. DMARDs: DMARDs (sykdomsmodifiserende antirevmatiske legemidler) som metotreksat kan brukes for å redusere betennelsen. Biologiske legemidler: I noen tilfeller kan biologiske legemidler som hemmer betennelsesstoffer (TNF-hemmere og IL-1 hemmere) være aktuelt.
Litteratur: Hospach C, 2019.
Early onseth enterocolitis (mutasjoner i IL-10R)
Early-onset enterokolitt er en sjelden genetisk sykdom som fører til alvorlig betennelse i tarmen hos barn. Sykdommen skyldes mutasjoner i genene IL10RA og IL10RB. Disse genene er viktige for å regulere immunforsvaret i tarmen. Mutasjonene fører til økt produksjon av betennelsesstoffer, blant annet TNF-α, som angriper tarmens slimhinne.
Symptomer
- Blodig diaré: Diaré med blod.
- Tarmsår: Sår i tykktarmen (kolon).
- Abscesser: Pussfylte byller i tykktarmen.
- Fistler: Unormale forbindelser mellom tarmen og huden eller andre organer.
- Sår i munnen: Smertefulle sår i munnhulen.
Behandling: Immunsuppressive legemidler: Legemidler som demper immunforsvaret kan forsøkes for å redusere betennelsen. Stamcelletransplantasjon: Hvis immunsuppressive legemidler ikke har effekt, kan transplantasjon av stamceller fra en frisk donor være et alternativ.
Litteratur: Glocker E-O, 2009.
Familiær kulde autoinflammatorisk syndrom (FCAS)
- Se kryopyrin assosiert sykdom (CPAS) nedenfor
Familiær middelhavsfeber (FMF)
Familiær Middelhavsfeber (FMF) er en arvelig sykdom som gir tilbakevendende episoder med feber og betennelse. Betennelsen kan ramme ledd, bukhinnen (peritonitt), lungehinnen (pleuritt) eller hjerteposen (perikarditt). Sykdomsårsak: FMF skyldes en mutasjon i et gen som heter MEFV. Dette genet er viktig for å regulere betennelsesreaksjoner i kroppen. Sykdommen arves autosomalt recessivt, som betyr at man må arve en kopi av det muterte genet fra begge foreldrene for å bli syk. Debutalder: Fire av 5 er under 20 år, to av tre under fem år ved første symptom. Debut etter 30-års alder er uvanlig. Forekomst: FMF er den vanligste monogene autoinflammatoriske sykdommen globalt, men er sjelden i Norge. Sykdommen er mest utbredt blant jøder, armenere, tyrkere, arabere og italienere (referanse: Mortensen SB, 2020).
Symptomer:
- Feber: Episoder med feber som kommer og går. Varigheten er 1-4 dager med uregelmessige intervall, ofte med 2-4 uker feberfrie perioder.
- Magesmerter: Sterke magesmerter på grunn av betennelse i bukhinnen.
- Leddbetennelse: Betennelse i store ledd som knær og ankler.
- Andre symptomer: Noen kan oppleve utmattelse, kvalme, appetittløshet, leddsmerter, brystsmerter, pustevansker og hoste før et anfall. I noen tilfeller kan det også oppstå hudutslett (referanse: Babaoglu H, 2020).
Diagnose: Diagnosen stilles basert på de typiske symptomene og kliniske funn. I forskning brukes det spesielle klassifikasjonskriterier for å stille diagnosen (referanse: Gattorno M, 2019).
Lignende tilstander/differensialdiagnoser: akutt infeksjon, barneleddgikt (oftest systemisk JIA), annen type periodisk febersyndrom / autoinflammatorisk sykdom, SLE, vaskulitt, blødende magesår (ulcus ventriculi) eller sterke magesmerter (akutt abdomen) av annen årsak, hereditært angioødem, akutt porfyri, tilbakevendende bukspyttkjertel-betennelse (pankreatitt), høyt blodfett (hyper-trigyceridemi), abdominal epilepsi, abdominal migrene.
Behandling: Kolkisin: Kolkisin er et legemiddel som reduserer betennelse og forebygger anfall. Ca. 90% har effekt. Biologiske legemidler: Hvis kolkisin ikke har tilstrekkelig effekt eller ikke tolereres (diare, magesmerte, utslett, lavt antall hvite blodlegemer og blodplater er blant mulige bivirkninger), kan biologiske legemidler som hemmer betennelsesstoffer (IL-1 hemmere, TNF-hemmere eller IL-6 hemmere) være aktuelt. Prognose: Behandlingen av FMF skal fortsette i mange år. Uten behandling kan sykdommen føre til en alvorlig komplikasjon som kalles AA-amyloidose. Dette er en tilstand der et protein avleires i ulike organer, spesielt nyrene, og kan føre til nyresvikt. Konsekvent behandling med kolkisin forebygger utvikling av amyloidose.
Litteratur: Kucuk A, 2014.
HIDS (Hyper IgD syndrom) (mevalonatkinase-mangel sykdom, MKD.
HIDS (Hyper IgD syndrom), også kjent som mevalonatkinase-mangel sykdom (MKD), er en sjelden arvelig stoffskiftesykdom. Sykdommen fører til tilbakevendende anfall med feber og betennelse.
Sykdomsårsak: HIDS skyldes en mutasjon i genet MVK, som er ansvarlig for å lage et enzym som kalles mevalonat kinase. Dette enzymet spiller en viktig rolle i kroppens stoffskifte. Mutasjonen fører til redusert aktivitet av mevalonat kinase, som forstyrrer stoffskiftet og fører til opphopning av mevalonsyre. Forekomst: IDS er mest utbredt i Nord-Europa, spesielt i Frankrike og Nederland. Sykdommen rammer oftest småbarn, vanligvis i løpet av første leveår.
Symptomer:
- Feber: Høye feberepisoder som varer i 4-5 dager.
- Utslett: Ulike typer hudutslett.
- Leddsmerter og betennelse: Smerter og betennelse i ledd (artritt).
- Magesmerter: Sterke magesmerter, ofte med oppkast og diaré.
- Hovne lymfeknuter: Hovne lymfeknuter på halsen.
- Hodepine: Intens hodepine.
- Forstørret milt: Miltens størrelse kan være økt.
- Sår: Sår i munnen og underlivet.
- Alvorlige former: I sjeldne tilfeller kan HIDS føre til alvorlige nevrologiske symptomer som synstap og hørselstap.
Diagnosen stilles basert på de typiske symptomene, kliniske funn og blodprøver. I blodprøver finner man forhøyede verdier av immunoglobulin D (IgD) hos de fleste pasientene, derav navnet Hyper IgD syndrom. I tillegg er CRP og senkning (SR) forhøyet. I forskning brukes det spesielle klassifikasjonskriterier for å stille diagnosen Eurofever/PRINO 2019 (referanse: Gattorno M, 2019).
Lignende tilstander (differensialdiagnoser): IgD økt i blod er forhøyet ved: Hodgkin-lymfom, sarkoidose, tuberkulose, aspergillose, ataxia-telangiectasia, HIV/AIDS.
Behandling: NSAIDs: Ikke-steroide antiinflammatoriske legemidler (NSAIDs) kan dempe smerter og betennelse. Steroider: Kortisonpreparater (steroider) kan brukes i perioder med mye betennelse. Biologiske legemidler: I noen tilfeller kan biologiske legemidler som hemmer betennelsesstoffer (TNF-hemmere eller IL-1 hemmere) forsøkes.
Forløp og prognose: Anfallene avtar ofte i voksen alder. I sjeldne tilfeller kan HIDS føre til amyloidose, en tilstand der et protein avleires i ulike organer.
Litteratur: Steichen et al. J Rheumatol 2009, van der Hilst & Frenkel. J Clin Rheum 2010. Mulders-Manders CM, 2015.
JMP Joint contractures – muscle atrophy – microcytic anemia – panniculitis-induced lipodystrophy
Sykdomsårsak: Sannsynligvis samme mutasjon som ved CANDLE syndrom (se «små barn» ovenfor), men JMP debuterer i voksen alder. Proteosom mutasjon. Symptomer: Ledd-kontrakturer, muskelsvinn, lav blodprosent / anemi, betennelse i fettvev (pannikulitt). Litteratur: Agarwal AK, 2010.
MKM (Mevalonatkinase-mangel)
- Vennligst se HIDS ovenfor.
Kryopyrin assosierte periodiske syndromer (CAPS) / Kryopyrin assosiert periodiske febersyndromer
Kryopyrin-assosierte periodiske syndromer (CAPS) er en gruppe sjeldne, arvelige sykdommer som fører til tilbakevendende episoder med feber og betennelse. Betennelsen kan ramme hud, ledd, øyne og hjernehinner.
Felles kjennetegn: Alle CAPS-sykdommene skyldes mutasjoner i et gen som heter NLRP3 (tidligere kalt CIAS1). Dette genet er viktig for reguleringen av betennelsesreaksjoner i kroppen. Mutasjonen fører til overproduksjon av et signalstoff som kalles interleukin-1 beta (IL-1β), som forårsaker betennelse.
Ulike former for CAPS: Dette er tre sykdommer med like kliniske trekk er de interleukin-1-assosierte, autoinflammatoriske sykdommene:
Det finnes tre hovedformer for CAPS:
- Familiær kulde-autoinflammatorisk syndrom (FCAS): Den mildeste formen for CAPS. Utløses av kuldeeksponering, for eksempel opphold i kaldt klima eller klimaanlegg. Symptomer er feber, leddsmerter og hudutslett.
- Muckle-Wells syndrom (MWS): En moderat form for CAPS. Symptomer er tilbakevendende episoder med feber, elveblestlignende utslett, leddbetennelse, øyebetennelse og utmattelse. Hørselstap og amyloidose kan forekomme som komplikasjoner.
- CINCA/NOMID syndrom: Den alvorligste formen for CAPS. Gir kronisk betennelse fra fødselen av, med symptomer som hudutslett, feber, leddbetennelse, øyebetennelse, hjernehinnebetennelse og forsinket utvikling.
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing.
Behandling: Behandling med biologiske legemidler som blokkerer IL-1 (anakinra, canakinumab) har god effekt hos mange. Behandlingen bør startes så tidlig som mulig i sykdomsforløpet for å forebygge komplikasjoner.
Litteratur: Tran TA, 2017.
Nakajo-Nishimuras sykdom (NNS)
Nakajo-Nishimuras sykdom (NNS) er en sjelden genetisk sykdom som fører til kronisk betennelse i kroppen. Sykdommen rammer oftest barn og kjennetegnes av hudutslett, feber, tap av fettvev og leddproblemer. Debutalder: Oftest omkring 2 års alder, men noen først i 6–12 års alder. Årsak: NNS skyldes en mutasjon i et gen som heter PSMB4. Dette genet er ansvarlig for å lage en viktig del av cellenes «resirkuleringssystem». Cellene våre inneholder proteasomer, som er små «maskiner» som bryter ned proteiner som ikke lenger trengs eller er skadet. Når PSMB4-genet ikke fungerer som det skal, påvirkes denne prosessen, og det kan føre til opphopning av avfallsstoffer og betennelse. Sykdommen arves autosomalt recessivt, som betyr at man må arve en kopi av det muterte genet fra begge foreldrene for å bli syk. Imidlertid har omtrent halvparten av de som får sykdommen ingen kjente tilfeller i familien.
Symptomer:
- Feber: Gjentatte feberepisoder.
- Hudutslett: Eksemlignende utslett.
- Tap av fettvev (lipodystrofi): Tap av fettvev under huden, spesielt i overkroppen.
- Leddstivhet (kontrakturer): Nedsatt bevegelighet i leddene.
- Lange fingre med fortykkede fingertupper (clubbing): Endringer i formen på fingrene.
Diagnosen stilles basert på de karakteristiske symptomene, kliniske funn og genetisk testing [mutasjon i PSMB4 (proteasom)]. Blodprøver kan vise tegn på betennelse, som forhøyet CRP og senkning, CK , gammaglobulin og ANA-test (med DNA og SSA-antistoff) og MPO-ANCA forekommer.
Behandling: Kortison: Kortisonpreparater (steroider) kan dempe betennelsen. DMARDs: DMARDs (sykdomsmodifiserende antirevmatiske legemidler) som metotreksat kan brukes for å redusere betennelsen. Biologiske legemidler: I noen tilfeller kan biologiske legemidler som hemmer betennelsesstoffer være aktuelt. Litteratur: Ohmura K, 2019.
Neonatal-Onset Multisystem Inflammatory Disease (NOMID) = Chronic Infantile Neurological Cutaneous Articular Syndrome (CINCA)
- Vennligst se CINCA ovenfor.
NLRP12-assosiert syndrom (nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain)
NLRP12-assosiert syndrom er en sjelden genetisk sykdom som fører til tilbakevendende episoder med feber og betennelse. Sykdommen ligner på CAPS (kryopyrin-assosierte periodiske syndromer), men skyldes en mutasjon i et annet gen, NLRP12.
Sykdomsårsak. NLRP12-genet er viktig for reguleringen av betennelsesreaksjoner i kroppen. Mutasjonen fører til en feil i dette genet, som kan føre til overdreven betennelse. Sykdommen arves dominant, som betyr at det er nok å arve en kopi av det muterte genet fra en av foreldrene for å bli syk. Imidlertid varierer sykdommens alvorlighetsgrad mye, selv innenfor samme familie. Forekomst: Tidlige barneår.
Symptomer: Symptomene ligner på CAPS (se ovenfor) og inkluderer:
- Feber: Høye feberepisoder som kommer og går, ofte med 3-4 ukers mellomrom. Anfallene kan vare i 2-10 dager.
- Leddsmerter: Smerter og stivhet i ledd.
- Muskelsmerter: Smerter og ømhet i muskler.
- Magesmerter og oppkast: Ubehag i magen og brekninger.
- Munnsår: Smertefulle sår i munnhulen.
- Hovne lymfeknuter: Forstørrede lymfeknuter.
- Hørselstap: Nedsatt hørsel.
Utløsende faktorer: Anfallene kan utløses av kuldeeksponering, for eksempel opphold i kaldt klima eller klimaanlegg.
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing. Genetisk testing kan påvise mutasjoner i NLRP12-genet.
Behandlingen fokuserer på å lindre symptomene og forebygge komplikasjoner. Det finnes ingen kur mot sykdommen.
Litteratur: Borghini S, 2011
NLRC4 inflammasom mutasjon
Mutasjoner i NLRC4-genet kan føre til en sjelden autoinflammatorisk sykdom som gir tilbakevendende feber og betennelse fra tidlig barnealder. NLRC4-genet er viktig for reguleringen av betennelsesreaksjoner i kroppen. Mutasjoner i dette genet kan føre til overdreven aktivering av inflammasomer, som er proteinkomplekser som spiller en rolle i immunforsvaret. Dette fører til økt produksjon av betennelsesstoffer, blant annet IL-1β.
Symptomer
- Tilbakevendende feber: Episoder med feber som kommer og går.
- Leddsmerter: Smerter og stivhet i ledd.
- Tarmbetennelse: Betennelse i tarmen som kan gi magesmerter, diaré og andre symptomer.
Komplikasjoner: En alvorlig komplikasjon ved NLRC4-inflammasommutasjon er makrofagaktiverings-syndrom (MAS). MAS er en livstruende tilstand med overdreven aktivering av immunforsvaret.
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing. Genetisk testing kan påvise mutasjoner i NLRC4-genet.
Behandling: Behandlingen fokuserer på å lindre symptomene og forebygge komplikasjoner. Det finnes ingen kur mot sykdommen.
Litteratur Canna SW, 2014).
PAPA syndrom (Akne, steril artritt eller abscess, pyoderma gangrenosum)
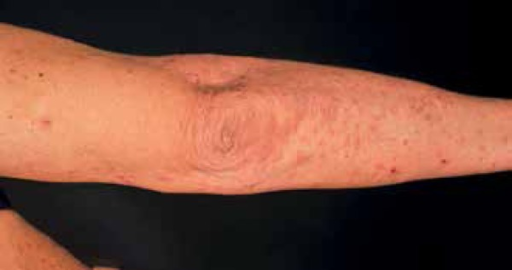
PAPA syndrom er en sjelden genetisk sykdom som fører til betennelse i hud og ledd.
Sykdomsårsak/genetikk: PAPA syndrom skyldes mutasjoner i et gen som heter PSTPIP1. Dette genet er viktig for reguleringen av betennelsesreaksjoner i kroppen. Mutasjoner i dette genet kan føre til overdreven betennelse. Debutalder: Hud-manifestasjoner begynner blant barn og unge voksne, artritt i løpet av 10-30 års alder.
Hudsymptomer:
- Patergi: Hudutslett og sår som oppstår etter små skader.
- Pyoderma gangrenosum: Smertefulle hudutslett med sår og betennelse.
- Alvorlig akne: Kviser og byller.
Leddsymptomer: Steril artritt: Betennelse i ledd uten tegn til infeksjon. Rammer oftest albue, kne eller ankel.
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing. Det kan være vanskelig å skille leddbetennelsen ved PAPA syndrom fra en infeksjon i leddet. Analyse av leddvæske kan hjelpe med å stille diagnosen.
Behandling: Ciclosporin A: Et legemiddel som demper immunforsvaret og kan ha god effekt på pyoderma gangrenosum (referanse: Kanameichi S, 2016). Biologiske legemidler: I noen tilfeller kan biologiske legemidler som hemmer betennelsesstoffer (IL-1-hemmere eller TNF-hemmere) forsøkes.
Litteratur: Genovese G, 2020.
PFAPA (Periodisk feber, aftøs stomatitt, pharyngitt of adenopati syndrome)

{kind=link}
PFAPA syndrom (Periodisk feber, aftøs stomatitt, faryngitt og cervikal adenitt) er den vanligste årsaken til tilbakevendende feber hos barn i Norge. Sykdommen kjennetegnes av regelmessige episoder med feber, munnsår, hovne lymfeknuter på halsen og sår hals (referanse: Rigante D, 2009). Årsaken til PFAPA syndrom er ukjent, men man tror at det skyldes en kombinasjon av genetiske og miljømessige faktorer. I motsetning til mange andre autoinflammatoriske sykdommer, som skyldes en mutasjon i et enkelt gen, tror man at PFAPA skyldes samspill mellom flere gener. (referanser: Esposito S, 2014; Stojanov S, 2011). Debutalder: Sykdomsdebut er før 5 års alder. Symptomene går vanligvis helt tilbake innen voksen alder.
Symptomer:
- Feber: Høye feberepisoder som kommer og går regelmessig, omtrent hver 2.-6. uke. Episodene varer i 3-6 dager.
- Munnsår (aftøs stomatitt): Smertefulle sår i munnhulen.
- Hovne lymfeknuter på halsen (cervikal adenitt): Forstørrede lymfeknuter på halsen.
- Sår hals (faryngitt): Rødhet og smerter i halsen.
- Andre symptomer: Hodepine og magesmerter kan også forekomme.
Diagnosen stilles basert på de typiske symptomene og kliniske funn. Det finnes ingen spesifikk test eller gen-test for å påvise PFAPA syndrom.
Behandling: Det finnes ingen behandling som kan kurere PFAPA syndrom, men symptomene kan lindres. Kortison: Kortisonpreparater (steroider) kan brukes i korte perioder for å dempe betennelsen, men risikoen for bivirkninger begrenser bruken (referanse: Quintana-Ortega C, 2020). Kolkisin: Kolkisin er et legemiddel som reduserer betennelse og kan ha effekt hos noen. Cimetidin: En type syrenøytraliserende medisin som kan redusere symptomene hos noen (referanse: Feder H, 1992). Biologiske legemidler: I noen tilfeller kan biologiske legemidler som hemmer betennelsesstoffer (IL-1-hemmere) forsøkes. Fjerning av mandler (tonsillektomi): Kirurgisk fjerning av mandlene kan vurderes hos noen pasienter.
Litteratur: Vanoni F, 2016.
SAVI (STING-assosiert vaskulopati med begynnelse i barndom).
SAVI er en sjelden og alvorlig genetisk sykdom som fører til betennelse i blodårene (vaskulitt) og lungene. Sykdommen starter i spedbarnsalderen og kjennetegnes av feber, lungesykdom og karakteristiske hudforandringer. SAVI skyldes en mutasjon i et gen som heter STING1. Dette genet er viktig for immunforsvaret og bidrar til å regulere kroppens reaksjon på virusinfeksjoner. Mutasjonen fører til overaktivering av STING1-proteinet, som igjen fører til overdreven produksjon av betennelsesstoffer.
Symptomer
- Feber: Gjentatte feberepisoder.
- Lungesykdom: Betennelse og arrdannelse i lungene (interstitiell lungesykdom).
- Hudbetennelse (vaskulitt): Betennelse i små blodårer i huden, spesielt på fingre, tær, ører og nese. Huden kan bli rød, hoven og smertefull, og det kan oppstå sår og vevsdød.
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing. Genetisk testing kan påvise mutasjoner i STING1-genet.
Behandling: Det finnes ingen effektiv behandling som kan kurere SAVI. Behandlingen fokuserer på å lindre symptomene og forhindre komplikasjoner.
- Medisiner: Ulike medisiner kan brukes for å dempe betennelsen og immunresponsen, inkludert kortison, immunsuppressive legemidler og biologiske legemidler.
- Støttebehandling: Støttebehandling er viktig for å håndtere lungesykdommen og hudforandringene. Dette kan inkludere oksygenbehandling, fysioterapi og hudpleie.
Prognose: SAVI er en alvorlig sykdom som kan føre til betydelig sykelighet og dødelighet. Alvorlighetsgraden av lungesykdommen og risikoen for infeksjoner er viktige faktorer som påvirker prognosen.
Litteratur: Wang Y, 2021)
Syklisk nøytropeni (hos barn)
Syklisk nøytropeni er en sjelden arvelig sykdom som fører til svingninger i antallet nøytrofile granulocytter i blodet. Nøytrofile granulocytter er en type hvite blodceller som spiller en viktig rolle i kroppens forsvar mot infeksjoner. Forekomst: Nye tilfeller (insidens) er 1-2/mill./år og forekomst (prevalens) 1-9/1 000 000. Sykdomsdebut før to års alder er typisk, men en adult/voksen form forekommer også. Sykdomsårsak: Sykdommen skyldes en mutasjon i et gen som heter ELANE. Dette genet er ansvarlig for produksjonen av et enzym som kalles nøytrofil elastase. Enzymet er viktig for nøytrofile granulocytters funksjon. Mutasjonen fører til en feil i dette enzymet, som forstyrrer produksjonen av nøytrofile granulocytter.
Symptomer
- Syklisk feber: Feber som kommer og går med jevne mellomrom, vanligvis hver 21. dag (14-35 dager). Feberen varer i 5-7 dager.
- Munnsår (stomatitt): Smertefulle sår i munnhulen.
- Tannkjøttbetennelse: Betennelse i tannkjøttet.
- Sår hals: Rødhet og smerter i halsen.
- Komplikasjoner: Personer med syklisk nøytropeni har økt risiko for infeksjoner, spesielt når antallet nøytrofile granulocytter er lavt. Alvorlige infeksjoner som blodforgiftning (sepsis) kan forekomme.
Diagnosen stilles basert på symptomene, blodprøver og genetisk testing. Blodprøver viser lavt antall nøytrofile granulocytter. Genetisk testing kan påvise mutasjoner i ELANE-genet.
Lignende tilstander (differensialdiagnoser): Idiopatisk (autoimmun) neutropeni: Ikke uvanlig med periodevis neutrofili på 500 – 1000/microL range, men med kronisk benignt forløp. Har vanligvis ikke de øvrige symptomene eller kronisk gingival infeksjon, andre autoimmune sykdommer (TRAPS, Hyper-IgD, FMF), Shwachman syndrom, «Lymfoproliferative disorders of large granular lymphocytes» (LDGL), primær immunsvikt vurderes når syklisk neutropeni og hepatosplenomegali forekommer.
Behandling: Granulocyttkolonistimulerende faktor (G-CSF): En medisin som stimulerer produksjonen av nøytrofile granulocytter. Infeksjonsbehandling: Antibiotika brukes for å behandle infeksjoner. Prognose: Sykdommen kan gå tilbake ved 30 års alder.
Litteratur: Newburger PE, 2013 (Neutropeni); Donadieu J, 2011.
TRAPS (Tumor-nekrose-faktor reseptor-assosiert periodisk syndrom)
TRAPS (Tumor-nekrose-faktor reseptor-assosiert periodisk syndrom) er en sjelden arvelig sykdom som gir tilbakevendende episoder med feber og betennelse. Sykdommen var tidligere kjent som Familiær Hiberniansk Feber. Årsak: TRAPS skyldes en mutasjon i et gen som heter TNFRSF1A. Dette genet er ansvarlig for å lage en reseptor for tumornekrosefaktor (TNF), et signalstoff som er viktig for betennelsesreaksjoner i kroppen. Mutasjonen fører til en feil i denne reseptoren, som igjen fører til overdreven betennelse. Forekomst: TRAPS er en svært sjelden sykdom. Man regner med at én person per million har sykdommen. Debutalder: Sykdomsstart i tidlig barnealder er vanligst med median debutalder 4,3 år. Imidlertid diagnostiseres ca. 10% av tilfellene hos personer over 30 års alder, da vanligvis med mildere varianter av sykdommen.
Symptomer: Episodene kommer hver 4-6 uke og varer 4-21 dager:
- Feber: Langvarige feberepisoder som kommer og går.
- Hudutslett: Rødt utslett som kan flytte seg rundt på kroppen.
- Magesmerter: Smerter i magen på grunn av betennelse i bukhinnen (peritonitt).
- Fordøyelsesproblemer: Diaré eller forstoppelse.
- Leddbetennelse: Betennelse i store ledd.
- Øyebetennelse: Betennelse i øynene (konjunktivitt, uveitt).
- Hevelse rundt øynene: Opphovning av øyelokkene.
- Muskelsmerter: Smerter og ømhet i muskler.
- Leggsmerter: Smerter i leggene på grunn av betennelse i muskelhinnen (fasceitt).
- Brystsmerter: Smerter i brystet.
- Hodepine: Intens hodepine.
Diagnosen stilles basert på de typiske symptomene, kliniske funn og genetisk testing. Genetisk testing kan påvise mutasjoner i TNFRSF1A-genet. I forskning brukes klassifikasjonskriterier Klassifikasjons-kriterier Eurofever/PRINO 2019 (referanse: Gattorno M, 2019).
Behandling: Biologiske legemidler som hemmer betennelsesstoffer (TNF-hemmere, IL-1-hemmere eller IL-6-hemmere) har ofte god effekt og kan redusere sykdomsaktiviteten, forebygge amyloidose og beskytte mot organskader (referanse: Cudrici C, 2020).
Litteratur
- Szekanecz Z, 2021
- Ter Haar NM, 2019
- Lidar M, 2017
- De Jesus AA, 2013
- Grans Kompendium i Revmatologi
- Helsebiblioteket/Pediatri veileder
- Du kan søke på sjeldne sykdommer via Orphanet (som også har en norsk informasjonsside)
- Mer om autoinflammatoriske sykdommer finner du også på NOMID Alliance (på engelsk)