

Definisjon
Inklusjonslegeme myositt (inclusion body myositis = IBM) er en betennelsesaktig muskelsykdom (myositt) som av ukjent årsak angriper eldre personer utvikler seg gradvis. IBM ble beskrevet som egen sykdom første gang i1978 (referanse: Carpenter, S, 1978). Lårmuskler og fingerbøyere (håndtrykk) er blant musklene som svekkes. Vevsprøve fra muskel er avgjørende for diagnosen. En har ingen gode legemidler mot denne typen myositt (referanse: McLeish E, 2022). IBM regnes som en del av myositt-sykdommene. Disse kan deles inn i fem grupper (referanse: Glaubitz S, 2020). 1) Polymyositt (PM), 2) Dermatomyositt (DM) (muskel- og hud-affeksjon), 3) Immunmediert nekrotiserende myopati (IMNM), inklusiv statin-indusert myopati, 4) Antisyntetase syndrom (lunger er angrepet og spesielle utslag i myositt-spesifikke antistoff foreligger) og 5) Inklusjonslegememyositt (IBM).
Forekomst
I Norge er det funnet at 3,3/100.000 har inklusjonslegememyositt (prevalens) (referanse: Dobloug C, 2015).. Sykdommen defineres dermed som en sjelden sykdom. Menn blir angrepet noe oftere enn kvinner. Vanlig alder ved diagnose er 50-70 år (referanse; Molberg Ø, 2016).
Det finnes en svært sjelden sykdomsform som skiller seg fra typisk IBM spesielt ved at den angriper tenåringer og unge voksne (referanse: Broccolini A, 2014). Denne hereditære (familiær/genetisk) IBM (hIBM) har dessuten en familiær forekomst (referanse: Ranque-Francois B, 2005). hIBM kalles også «Distal myopati med rimmed vacuoles» og er en genetisk sykdom. Denne formen er ikke nærmere omtalt her.
Klassisk IBM kalles også sporadisk inklusjonslegeme myositt og er regnet som en av de vanligste muskelsykdommene hos personer over 50 år (referanse: de Paepe B, 2018).
Symptomer
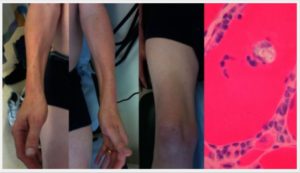
Fordi symptomene utvikler seg langsomt tar det ofte 5-6 år før en endelig diagnose stilles (referanse: Dobloug GC, 2014).
Lårmusklene svekkes gradvis slik at en får problemer med å reise seg fra huk-sittende og etter hvert fra stolen. Vansker med å gå opp trapper er også vanlig.
Underarmer, fingre og føtter. Muskler i underarmer og legger blir ofte tynnere og svakere etter hvert. Håndkraften (håndtrykk) svekkes og gangfunksjonen påvirkes med økt fall-tendens.
Asymmetri. Svakheten trenger ikke være lik i begge kroppshalvdeler (asymmetrisk affeksjon), selv om forandringene forventes på begge sider i forløpet.
Svelgemuskler. Enkelte får redusert svelgemuskel funksjon og svelgeproblemer (dysfagi). Det kan medføre at føde kommer ned i lunger (aspirasjon) og forårsaker lungebetennelser.
Polynevropati merkes med økende nummenhet og følelsesløshet, oftest i føtter og hender.
Undersøkelser
Inklusjonslegeme myositt har etter hvert typiske kjennetegn, men de er vanskelige å fastslå tidlig i sykdomsforløpet.
Sykehistorien kartlegger aktuelle symptomer (se ovenfor) med vekt på utvikling av svikt i underarmer, hender, legger og føtter, men også i lårmuskler. Svelgeproblemer etterspørres. Noen plages med muskelsmerter og nummenhet (polynevropati).
Klinisk undersøkelse observer muskelstørrelse i ben og armer. En kan se tynne armer og legger (distal muskelatrofi) (illustrasjonen ovenfor). Muskelstyrken vurderes: Gange på hæler og tær kan være vanskelig, og det kan være problematisk å reise seg fra huk eller fra stol uten støtte. og svakhet: Finger fleksorer, gripe-funksjon (oftest ikke-dominant hånd). Fingrene kan være bøyde (fleksjonsstilling). –Fysioterapeut kan bruke standardiserte tester for muskelstyrke, noe som kan være viktige verktøy i oppfølgingen, særlig for å vurdere ev sykdomsutvikling over tid eller om behandlingsmål oppnås. Fysioterapeuter tester både direkte muskelstyrke og utholdenhet.
Blodprøver viser ofte at kreatin kinase (CK) er forøket til 2-5 ganger øvre normal verdi. Andre prøver tas for langt på vei å utelukke andre tilstander og omfatter CRP, SR, hvite blodlegemer (leukocytter med differensialtellinger), blodplater (trombocytter), elektrolytter, lever-, nyre-, og thyreoidea-funksjonsprøver, glukose.
Ingen klassiske antistoffer forventes å foreligge. Anti-cytosolic 5′-nucleotidase 1A (cN-1A) har i en studie vist sensitivitet (evne til å fange opp IBM) på 50% (referanse: Felice KJ, 2018), men i andre kohorter noe lavere, ca. 30%. Antistoffet er dessverre ikke spesifikt for IBM og ses også ved andre tilstander, selv uten muskel-manifestasjoner (referanse: Lloyd TE, 2016). cN-1A er tilgjengelig for analyse (blot) blant myositt-spesifikke antistoff.
EMG/nevrografi kan vise myopati forandringer
Bildediagnostikk. omfatter MR av muskler, oftest lårmuskler. Forandringer som er ganske karakteristiske (referanse: Tasca G, 2015). En ser tydelig tap av muskulatur (atrofi) og fettinfiltrasjon. Typisk lokalisasjon er på strekk-siden av lårmuskler, særlig quadriceps- muskler nær kneet (distalt på låret). Ofte en gradient med økende forandringer nærmest kneet. Også sartorius-musklene angripes ofte, mens recutus femoris muskelen angripes ikke hos alle. Muskler bak låret (fleksor-muskler) angriper mer variabelt og kan være normale. Røntgen av spiserør viser redusert svelgefunksjon hos ca. 60%. Ultralyd-undersøkelse av muskulatur kan vise samme fordeling av atrofi som ved MR (referanse: Leeuwenberg KE, 2020).
Vevsprøve (biopsi) er ofte avgjørende for diagnosen. En ser både såkalte «degenerative og inflammatoriske trekk» med endomyseal invasjon av CD8+ lymfocytter, cytoplasmatiske og intranukleære inklusjoner. Amyloid i muskelfibre (krystall-fiolett-farging) (”Alzheimer i musklene”). Lysmikroskopi: Vakuoler, intra-nukleære og intra-cytoplasmatiske inklusjoner. Elektronmikroskopi: Mikrotubulære filamenter i inklusjonene. Disse blir farget med Kongo rødt, thioflavin og krystallfiolett (indikerer amyloidose) (referanse: Tasca G, 2015). Typiske funn ved biopsi er dermed Vakuoler (“rimmed vacuoles”, “inclusion bodies”), endomyseal inflammasjon, intracellulære amyloid-depoter og tubulofilamenter.
Diagnose
For diagnosen er sykehistorie, alder ved debut, sykdomsforløp og undersøkelser avgjørende (se ovenfor). En kan også bruke diagnostiske kriterier:
Diagnostiske kriterier
Adaptert etter “The ENMC IBM research diagnostic criteria 2011″ (referanse: Rose MR. 2011). Spesifisitet 99%, sensitivitet 57%.
| Klinikk og laboratorium | Klassifikasjon | Patologiske funn |
|---|---|---|
| Varighet > 12 måneder. Alder ved debut > 45 år. Kne-ekstensjon svakere enn hofte-fleksjon og/eller finger fleksjon svakhet > skulder abduksjon svakhet. CK ikke høyere enn 15× øvre referanse-grense | Klinikk‐patologisk definert IBM | Alle de følgende: Endomyseal inflammatoriske infiltrater- Rimmed vacuoles (inklusjonslegemer) Protein akkumulering a eller 15‐ to 18‐nm filamenter |
| Varighet > 12 måneder. Alder ved debut > 45 år. Kne-ekstensjon svakere enn hofte-fleksjon og/eller finger fleksjon svakhet > skulder abduksjon svakhet. CK ikke høyere enn 15× øvre referanse-grense | Sannsynlig / mulig IBM | En eller flere, men ikke alle av de følgende: Endomyseal inflammatoriske infiltrater. Rimmed vacuoles (inklusjonslegemer). Protein akkumulering eller 15‐ to 18‐nm filamenter |
Lignende tilstander/ differensialdiagnoser
- Amyloidose
- Annen myositt
- Nevrologisk sykdom, inklusiv amyotrofisk lateralsklerose (ALS)
- Non-inflammatorisk myopati (nevromuskulære sykdommer, lagringssykdommer og andre årsaker til muskelsmerter.
- Sarkoidose (med myopati)
Behandling
Alle anbefales fysioterapi, ergoterapi og egentrening for å redusere risiko for fall og skader, samt psykologisk støtte. Fysisk trening kan bedre mitokondrie-funksjon (cellenes kraftverk), blodsirkulasjonen, muskelvekst og redusere muskelbetennelse. Tilpasset kosthold for god ernæring optimal kroppsvekt vektlegges, men det finnes ingen spesielle tilskudd som er vist å påvirke sykdomsforløpet.
Ved svelgeproblem (dysfagi) vurderes behov for faryngo-øsofagal dilatasjon eller criko-faryngeal myotomi (referanse: Oh TH, 2008).
Medikamenter har dessverre liten eller ingen effekt og bivirkninger kan overgå nytten, men respons på behandling er forskjellig fra person til person. Iblant gjøres et behandlingsforsøk over 6 måneder. I så fall gjøres kartlegging av sykdommen og behandlingsmål skisseres på forhånd. Etter effektvurdering blir det vurdert om videre medikamentell behandling er aktuelt. Hvis behandlingsforsøk med immundempende medikamenter gjøres, er doseringen individuell. Et regime som er brukt er: Prednisolon 0,5–1mg/kg/dag første uken, deretter reduksjon slik at dosen er 10mg/d etter noen uker. Denne dosen opprettholdes i inntil 6 måneder. En må være oppmerksom på at det generelt er usikker effekt av kortikosteroider, selv ok CK verdiene blir bedre. Prednisolon kombineres ofte med metotreksat 7,5mg – 25 mg/uke over 3 – 6 måneder eller Imurel 1,5 – 2,5 mg/dag. Hvis ikke effekt på muskelstyrke og progresjon etter 6 måneder, bør en vurdere å avslutte behandlingen. Det er ikke sikker virkning av Sendoxan, Leukeran, azathioprin (Imurel), Ciclosporin A) eller metotreksat hos de fleste.
Immunglobulin (IVIG, Oktagam) intravenøst kan forsøkes ved alvorlig affeksjon av øsofagus og svelgevansker (referanse: Cherin, P, Pelletier, S, Neurology 2002). Noen kan ha forbigående effekt på svelgevansker (Dobloug C, 2012), men generelt er behandlingseffekten av IVIG ikke godt dokumentert (Glaubitz S, 2020). Det pågår utprøving av nye legemidler for IBM (Naddaf E, 2018).
Prognose
Sykdommen er ikke dødelig, men medfører økende behov for hjelpemidler. Forløpet er individuelt og antakelig også avhengig av om trening og andre tiltak gjennomføres. Gjennomsnittlig klarer en seg uten gang-hjelpemidler i 7,5-10 år fra symptomstart. Deretter brukes for eksempel staver eller stokk. Etter 13-15 år vil de fleste ha behov for transport i rullestol (referanse: Greenberg SA, 2019).
Retningslinjer
Litteratur
- Panginikkod S, 2022
- Naddaf E, 2018
- Molberg Ø, 2016
- Hilton-Jones , 2016
- Dobloug GC, 2015
- Grans Revmakompendium