

Definisjon
Marfans syndrom er en arvelig sykdom som påvirker bindevevet i kroppen. Bindevev er en viktig del av kroppens støttestruktur og finnes i blant annet hud, sener, leddbånd, blodårer og øyne. Sykdommen skyldes en feil (mutasjon) i genet som lager proteinet fibrillin-1. Dette proteinet er en viktig byggestein i bindevevet. Feilen i genet gjør at bindevevet blir svakere og mer elastisk enn normalt. Marfans syndrom er en medfødt tilstand, noe som betyr at man er født med den. Den skiller seg fra autoimmune bindevevssykdommer, som for eksempel revmatoid artritt, som skyldes at kroppens immunsystem angriper sitt eget bindevev.
Årsak
Sykdommen skyldes en arvelig defekt (misfolding) i proteinet fibrillin-1som kodes av genet FBN1 på kromosom15. Omtrent 25% er spontanmutasjoner uten familiedisposisjon (referanse: Store Norske Leksikon, A Heiberg 2009).
Forekomst
Marfans syndrom er en sjelden sykdom. Det anslås at omtrent 1 av 5000 personer har sykdommen. Likevel regnes den som en av de vanligste arvelige sykdommene som påvirker muskel- og skjelettsystemet (referanse: Gray JR, 1994).
Sammen med en vaskulær form (som angriper blodårer) av Ehlers-Danlos syndrom, Loeys-Dietz syndrom og arvelig thorakalt aortaaneurisme (HTAD), inngår sykdommen blant de arvelige” Marfans-lignende” sykdommene.
Symptomer
Marfans syndrom kan gi en rekke ulike symptomer, og symptomene varierer fra person til person. Noen vanlige symptomer er:
Skjelettsystemet:
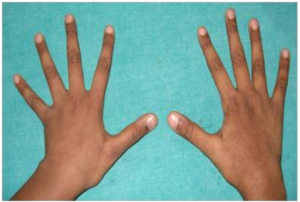
- Høy vekst (referanse: Erkula G, 2002).
- Lange armer, ben (dolichostemomeli), fingre og tær (arachnodaktyli) (referanse: Lindsey J, 1998).
- Skjev rygg (skoliose)
- Brystben-deformiteter [fuglebryst (pectus carniatum) eller traktbryst]
- Overbevegelige ledd (hypermobilitet) (referanse: Kaissi AA, 2013).
Øyne:
- Nærsynthet (myopi)
- Skjeve hornhinner (astigmatisme)
- Linseluksasjon (linsen i øyet er forskjøvet/ektoptisk linse)
- Grønn stær (glaukom)
- Grå stær (katarakt) (referanse: Zech J-C, 2020)
Hjertet og blodårer:
- Uregelmessig hjerterytme
- Hjerteklaff-lekkasje
- Utvidet hovedpulsåre (aortaaneurisme)
- Aortadisseksjon (rift i hovedpulsårens vegg) (referanse: Rybczynski M, 2010).
Lunger: Punktert lunge (pneumotoraks), vanligst i lungenes overlapper (øvre del).
Hud: Strekkmerker (striae)
Diagnosen
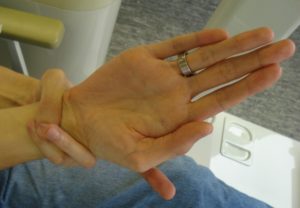
Diagnosen Marfans syndrom stilles på bakgrunn av symptomer, kliniske funn og genetiske undersøkelser.
Legen vil undersøke pasienten og se etter typiske tegn på sykdommen. Det finnes også kriterier (Ghent-kriteriene) som brukes for å stille diagnosen. Disse kriteriene tar hensyn til både symptomer, familiehistorie og genetiske funn.
I noen tilfeller kan det være nødvendig med genetisk testing for å påvise mutasjonen i FBN1-genet.
Ofte brukes også kriterier for diagnosesetting (2010 Ghent) som skiller mellom de som har Marfans sykdom i nærmeste familie og de uten arvelig disposisjon:
Uten arvelig disposisjon:
- Aortarot utvidet (Z-skår ≥2) og ektopisk linse
- Aortarot utvidet (Z-skår ≥2) og FBN1 mutasjon (gentest)
- Aortarot utvidet (Z-skår ≥2) og systemisk skåring* >7 punkter (spesielt skåringssystem*)
- Ektopisk linse og FBN1 mutasjon med kjent aortaaffeksjon
Med familie med Marfans sykdom:
- Ektoptisk linse
- Systemisk skåring* Aortarot utvidet ≥7
- Aortarot utvidet (Z-skår ≥2)
*Skår: Pectus carniatum=2 (pes excavatum eller assymmetri=1), Malleol-deformitet (ankel)=2 (plattfot=1), Dural ektasi=2, Protrusio acetabuli=2, Dys-proporsjonal armlengde =1, Skoliose eller kyfose=1, Redusert albue ekstensjon=1, Ansiktsskjelett forandringer= 1, Striae i huden=1, myopi=1, Mitralprolaps =1.
Gamle kriterier: major kriterier (minst 4):
- Redusert proporsjon overkropp/underkropp ratio (0,85 vs 0,93 hos normale)
- Arachnodactyli (fingre, tær), positiv tommel-håndledd-test)
- Skoliose mer enn 20 grader eller spondylolistese
- Medial malleol-feilstilling som gir pes planus (plattfot)
- Redusert ekstensjon i albuer (<170 grader)
- Pectum carniatum (fuglebryst) (Differensialdiagnose: Morquio syndrome, Noonan syndrome, Trisomi 18, Trisomi 21, Homocystinuri, Osteogenesis imperfecta, multiple lentigines syndrom, and Sly syndrome)
- Pectus excavatum (traktbryst) som krever kirurgi
- Protrusio Acetabuli (Differensial diagnoser: Pagets syndrom, revmatoid artritt, Bekhterevs sykdom, Osteomalasi)
Lignende sykdommer (differensialdiagnoser)
Det finnes flere andre sykdommer som kan ligne på Marfans syndrom. Noen av disse er (alfabetisk):
- Arvelig thorakalt aortaaneurisme (HTAD): HTAD kan ligne Marfans syndrom ved at begge tilstandene disponerer for utvidelse (aneurisme) og rift (disseksjon) i hovedpulsåren (aorta), men HTAD er ofte mer spesifikt for aorta og mangler vanligvis de andre karakteristiske trekkene ved Marfans syndrom, som skjelett- og øyeproblemer. HTAD kan skyldes mutasjoner i ulike gener, inkludert gener som også er involvert i Marfans syndrom (som FBN1), men også andre gener som ACTA2 og MYH11.
- Ehlers-Danlos syndrom (EDS): EDS kan ligne Marfans syndrom ved å forårsake løse ledd (hypermobilitet), tøyelig hud og problemer med blodårene, men EDS er en gruppe av flere ulike arvelige bindevevssykdommer med varierende alvorlighetsgrad og spesifikke trekk. EDS kjennetegnes ofte av mer uttalt hudtøyelighet og -sårbarhet enn ved Marfans syndrom, og involverer sjeldnere linseluksasjon. Vaskulær EDS er den formen som mest ligner Marfans på grunn av risikoen for arterielle rupturer.
- Ektoptisk linse syndrom (ELS) Ektoptisk linse syndrom (ELS), familiær thorakalt aorta aneurisme og disseksjon syndrom (FTAAD/FTAA). Denne tilstanden kombinerer linseluksasjon (som ved Marfans syndrom) med en økt risiko for aortaaneurisme og disseksjon, og kan derfor ligne Marfans syndrom. Imidlertid kan årsaken være mutasjoner i gener som ikke er FBN1 (genet som er mutert ved klassisk Marfans), og andre karakteristiske trekk ved Marfans syndrom er kanskje ikke til stede.
- Hypermobilitetssyndromet kjennetegnes av løse ledd og kan derfor ligne Marfans syndrom. Imidlertid har personer med hypermobilitetssyndromet vanligvis ikke de samme alvorlige hjerte- og øyeproblemene som ofte sees ved Marfans syndrom.
- Loeys-Dietz syndrom (LDS) (TGF-beta receptor 1 og 2 genmutasjoner, prøve sendes OUS, UUS): LDS kan ligne Marfans syndrom svært mye, da begge tilstandene involverer problemer med aorta (aneurisme og disseksjon), skjelettforandringer og noen ganger øyeproblemer. LDS kjennetegnes imidlertid ofte av mer uttalte ansiktsforandringer (f.eks. bredt ansikt, hypertelorisme (økt avstand mellom øynene), spaltet uvula) og arterielle tortuosities (svingete arterier) som ikke er like vanlige ved Marfans syndrom. LDS skyldes mutasjoner i gener som koder for TGF-beta reseptorer (TGFBR1 og TGFBR2).
- Menkels sykdom (defekt i kobber-metabolisme): Menkes sykdom er en sjelden, arvelig kobbermetabolismeforstyrrelse som kan forårsake løs hud, sprøtt hår og vaskulære problemer, som kan ligne noen aspekter ved Marfans syndrom. Menkes sykdom har imidlertid distinkte kjennetegn som kramper, utviklingsforsinkelse og karakteristiske hårforandringer (kinky hair) som ikke sees ved Marfans syndrom.
- Pseudoxanthoma elasticum (PXE): PXE er en genetisk bindevevssykdom som hovedsakelig påvirker hud, øyne og blodårer. Hudforandringene (små, gule papler) og øyeproblemene (angioid streaks) kan forveksles med noen av symptomene ved Marfans syndrom, men PXE involverer ikke de typiske skjelettforandringene eller de samme hjerteproblemene som ved Marfans syndrom.
- Stickler syndrom (hereditær arthro-oftalmopati). Stickler syndrom er en gruppe arvelige tilstander som påvirker bindevevet, spesielt i øyne, ører, ledd og ansikt. Noen trekk ved Stickler syndrom, som myopi (nærsynthet), leddproblemer og ansiktsforandringer, kan overlappe med Marfans syndrom. Imidlertid er ansiktsforandringene ved Stickler syndrom ofte mer karakteristiske (f.eks. flatt ansikt, liten nese), og det er vanlig med hørselstap, noe som ikke er typisk for Marfans syndrom.
- Weill-Marchesani syndrom (WMS). WMS er en sjelden, arvelig bindevevssykdom som kjennetegnes av kortvoksthet, korte fingre og tær (brakydaktyli), linseluksasjon og myopi. Linseluksasjonen kan forveksles med den som sees ved Marfans syndrom, men de andre karakteristiske trekkene ved WMS, som kortvoksthet og brakydaktyli, skiller den fra Marfans syndrom, som vanligvis involverer høy vekst og lange lemmer.
Behandling
Det finnes ingen behandling som kan kurere Marfans syndrom. Behandlingen har som mål å lindre symptomer og forebygge komplikasjoner, spesielt fra hjerte og blodårer.
- Medisiner: Betablokkere kan redusere belastningen på hjertet og blodårene.
- Kirurgi: Operasjon kan være nødvendig for å reparere aortaaneurismer eller hjerteklaffer. Gjennomsnittsalder for operasjon er 32 år.
- Ergoterapi: Stabiliserende ortoser og tilpasset fottøy.
- Livsstilsråd: Det er viktig å unngå aktiviteter som øker belastningen på hjerte og blodårer, for eksempel intensiv trening og kontaktsport.
Behandlingen er ellers avhengig av eventuelle komplikasjoner fra øyne, ledd og columna / thoraks. Ingen helbredende (gen-terapi) behandling er tilgjengelig.
Regelmessige kontroller: Personer med Marfans syndrom trenger regelmessige kontroller hos lege, spesielt hos hjertespesialist. Kontroller kan omfatte MR-angiografi fremstiller de viktigste pulsårene fra og med hodet, halsen, brystet, mageområdet og bekkenet. Også med ultralyd Doppler / ekkokardiografi kan diameter på hovedpulsåren ved hjertet og aortabuen måles. Hvis pulsåren utvider seg, gjøres målinger minst hvert halvår, ellers årlig i en lengre periode. Også ved CT-undersøkelse kan hele hovedpulsåren fremstilles. Undersøkelsen gjøres ikke for ofte på grunn av røntgenstråling.
Prognose
Prognosen for personer med Marfans syndrom er blitt betydelig bedre de siste årene, takket være bedre diagnostikk og behandling.
Litteratur
- Kompetansesenter, Sunnås Sykehus
- Salik I, 2023
- Dietz HC, 2001
- Marfan-foreningen i Norge
- Barstow C, 2015
- Grans Kompendium i Revmatologi