


Definisjon
Primær immunsvikt er en gruppe sykdommer forårsaket av forskjellige genfeil. Disse feilene fører til at immunforsvaret ikke fungerer som det skal, noe som gjør kroppen mer utsatt for infeksjoner. Symptomene oppstår vanligvis i barnealder, med mer enn 60 % av tilfellene før 5-årsalderen. Omtrent 20 % debuterer først i voksen alder. Noen ganger kan immunsvikten føre til at kroppens eget immunsystem angriper friskt vev. Dette kalles autoimmunitet og kan ytterligere svekke immunforsvaret. Noen personer med primær immunsvikt har også autoimmune sykdommer. Primær immunsvikt må ikke forveksles med sekundær immunsvikt, som oppstår som følge av andre faktorer som immundempende legemidler, diabetes, alvorlig nyresykdom, HIV-infeksjon, kreft og høy alder.
Forekomst
Man regner med at det finnes omtrent 400 personer med alvorlig primær immunsvikt i Norge. Data fra det europeiske registeret for immunsviktsykdommer (ESID) viser at det finnes rundt 500 forskjellige typer primær immunsvikt. Registeret inneholder informasjon om ca. 30 000 tilfeller, fordelt slik:
| Primære immundefekter | Andel |
| Antistoff-relaterte sykdommer | 50,4% |
| Kombinerte immundefekter | 10,3% |
| Fagocytose-defekter | 8,2% |
| Immun dysregulasjon | 5,9% |
| Autoinflammatoriske sykdommer | 3,0% |
| Uklassifiserte immundefekter | 1,4% |
| Benmargsassosierte | <1% |
| Andre definerte primære immundefekter | 15,1% |
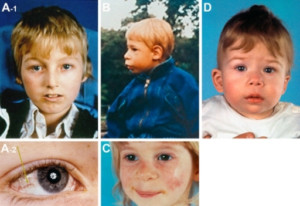
Symptomer
Vanlige symptomer på primær immunsvikt er hyppige infeksjoner, som for eksempel: Lungebetennelser, bronkitter, bihulebetennelser, ørebetennelser, meningitt og hud-infeksjoner.
Andre symptomer er febertilstander uten infeksjon, allergi-lignende plager, redusert hormon/kjertel-funksjon og vekstforstyrrelser hos barn, tarmplager og kvalme. Noen fremviser tegn på autoimmun sykdom i form av utslett og hovne lymfeknuter.
Undersøkelser
For å stille diagnosen primær immunsvikt vil legen gjøre en grundig vurdering av sykehistorie, kliniske funn og resultater fra ulike undersøkelser:
Sykehistorie: Legen vil spørre om familiehistorie med arvelige sykdommer, tidligere sykdommer, medisinbruk, autoimmune sykdommer, kreft, veksthemming og diaré.
Klinisk undersøkelse: Legen vil undersøke huden for utslett, leddene for hevelse og smerter, og lymfeknutene for forstørrelse. I tillegg vil legen undersøke lungene, hjertet og andre indre organer, samt nervesystemet, syn og hørsel. Tegn på SLE (lupus), revmatoid artritt (leddgikt) og diabetes bør utelukkes (noe økt forekomst).
Blodprøver. Ulike blodprøver kan tas for å vurdere immunforsvaret, inkludert celletellinger, måling av immunglobuliner ( IgG, IgM, IgA) og tester for autoimmunitet (anti-nukleære antistoff (ANA) og anti-CCP), s-elektroforese, albumin, virus: HIV, hepatitt, CMV, EBV.
Bildediagnostikk. CT av brystkasse og mage kan være aktuelt for å visualisere indre organer.
Vevsprøve (biopsi): I noen tilfeller kan det være nødvendig å ta vevsprøver fra benmarg (evt. med supplerende flowcytometri), lymfeknuter, hud eller slimhinner for å undersøke cellene nærmere.
Spesialtester: Mer spesialiserte tester for å vurdere ulike deler av immunforsvaret kan være aktuelt, for eksempel tester for T-cellesvikt, B-cellesvikt, fagocytosedefekter og komplementdefekter. Genetiske undersøkelser (genotyping) kan også være aktuelt.
Diagnosen immunsvikt stilles oftest av en barnelege, immunolog eller hematolog.
Lignende tilstander (differensialdiagnoser)
Det er viktig å utelukke andre tilstander som kan ligne på primær immunsvikt, for eksempel:
- Sekundær immunsvikt
- Autoimmune sykdommer (f.eks. SLE (lupus), revmatoid artritt (leddgikt))
- Infeksjoner med uvanlige mikrober (opportunistiske infeksjoner)
Noen spesifikke immunsvikt–diagnoser
- Antistoffsvikt: Brutons agammaglobulinemi, X-bundet agammaglobulinemi, hypogammaglobulinemi, CVID (Common Variable Immundeficiency Disease), IgG subklassedefekter (selektiv IgA-mangel, Hyper-IgM syndrom).
- T-celle og kombinert B- og T-cellesvikt: Alvorlig kombinert immunsvikt (SCID = severe combined immunodeficiency), Omenn syndrom, RAG 1/ RAG 2 defekt, Retikulær dysgenesi X-bundet SCID, IL2RG Jak 3 mangel, ZAP70 mangel, Wiscott Aldrich syndrom, X-bundet trombocytopeni, Di George anomali (velofacialt syndrom eller CATCH 22), Ataxia telangiectasia.
- Fagocyttdefekter: Kronisk granulomatøs sykdom/(CGD), Glucose-6-Fosfat dehydrogenase mangel, Myeloperoxidase mangel, Interferon gamma receptor defekt, Leukocytt adhesjon defekt (LAD). Medfødt neutropeni, Severe congenital neutropenia, (SCN), Syklisk neutropeni (Kostmann syndrom), Idiopatisk neutropeni, Schwachman-Diamond syndrom
- Komplement defekter: C1 inhibitor defekt (Hereditært angioødem). C2, C4, C5, C6, C8 mangel og andre komplementdefekter
- Immunsvikt assosiert med andre syndromer: Hyper IgE syndrom, Kronisk mukocutan candidiasis inkludert APECED/APC-1/Whitaker syndrom, autoimmun-polyendokrinopati-candidiasis-ectodermal dysplasi. Ivemark syndrom (hjertefeil, situs inversus og manglende milt ved fødselen), X-bundet lymfoproliferativt syndrom (Duncan syndrom)
- MBL-mangel: MBL (Mannosebindende lektin) er et protein som spiller en viktig rolle i kroppens immunforsvar. Det bidrar til å bekjempe infeksjoner ved å gjenkjenne og binde seg til skadelige mikroorganismer som bakterier og virus. Noen personer har naturlig lave nivåer av MBL i blodet, noe som kalles MBL-mangel. Dette kan øke risikoen for å utvikle alvorlige infeksjoner, spesielt meningokokksykdom. MBL-mangel kan også øke risikoen for blodpropp og påvirke forløpet av autoimmune sykdommer som bindevevssykdommer og vaskulitter. MBL-mangel er relativt vanlig, med en forekomst på omtrent 5 % i befolkningen. De fleste med MBL-mangel har ingen symptomer, men de kan være mer utsatt for alvorlige infeksjoner. MBL-mangel påvises ved en enkel blodprøve. Det finnes ingen spesifikk behandling for MBL-mangel, men fokus rettes mot å forebygge infeksjoner og behandle eventuelle komplikasjoner. Forebyggende tiltak kan inkludere vaksiner, god hygiene og rask behandling av sår. Ved lave nivåer av immunglobulin-G (IgG) i blodet, kan behandling med immunglobuliner være aktuelt (referanse: K Takahashi, 2011)
Behandling
Behandlingen av primær immunsvikt avhenger av hvilken type immunsvikt man har.
Antistoffsvikt: Personer med antistoffsvikt kan behandles med immunglobuliner (antistoffer) som gis intravenøst eller subkutant (under huden). Dette er en livslang behandling som gis regelmessig for å styrke immunforsvaret. Forebyggende behandling med antibiotika kan også være nødvendig. I noen tilfeller kan stamcelletransplantasjon være et alternativ.
T-cellesvikt: T-cellesvikt er generelt vanskeligere å behandle. Thymustransplantasjon kan være et alternativ for noen pasienter med DiGeorge syndrom.
Kombinert immunsvikt (SCID): Benmargstransplantasjon kan være livreddende for personer med SCID. Genterapi er en lovende ny behandlingsform som forventes å bli tilgjengelig i fremtiden.
Litteratur
- Angel A, 2023
- Bonilla FA, 2016
- Kersseboom R, 2011
- Senter for sjeldne diagnoser
- Grans Kompendium i Revmatologi