

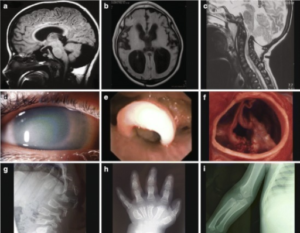
Definisjon
Lagringssykdommer er en gruppe sjeldne, genetiske sykdommer. De tilhører ofte en større gruppe kalt lysosomale lagringssykdommer (LSD). Felles for disse sykdommene er en feil i stoffskiftet som fører til at store molekyler (makromolekyler) hoper seg opp i kroppens organer. Disse molekylene fortrenger normalt vev, og reduserer dermed organenes funksjon gradvis. Lagringssykdommer kan ramme ulike organer, men ofte er lever, milt og hjerte affisert. I noen tilfeller er også hjernen involvert. De fleste lagringssykdommer oppdages i tidlig barnealder. Noen kan i starten ligne på revmatiske sykdommer eller bindevevssykdommer (referanse James RA, 2016). Mitokondriesykdommer som også regnes blant de metabolske er omtalt på en egen side.
Sykdomsårsak
Lagringssykdommer skyldes mangel på viktige enzymer i cellenes lysosomer. Lysosomer er cellens «renholdsverk» som bryter ned avfallsstoffer. Når et enzym mangler, kan ikke visse stoffer brytes ned, og de hoper seg opp i cellene. Hver sykdomstype er vanligvis knyttet til mangel på ett spesifikt enzym, noe som fører til unike symptomer.
Forekomst
Lysosomale sykdommer rammer omtrent 1 av 7000 nyfødte. Enkelte diagnoser er mer utbredt i spesifikke etniske grupper eller geografiske områder.
Symptomer
Lagringssykdommer varierer i alvorlighetsgrad og hvilke organer som rammes. Noen debuterer allerede før fødsel, mens andre viser seg i barne- eller voksen alder.
Vanlige symptomer hos barn:
- Forsinket utvikling
- Vekstforstyrrelser
- Økende størrelse på lever og milt
- Økende mageomfang
- Gradvis hjertesvikt
- Påfallende tung pust ved belastning
- Væske i bena
- Kalde, blålige hender
- Nedsatt hørsel og syn
- Kramper (epilepsilignende)
Undersøkelser
Følgende undersøkelser kan være aktuelle:
- Prenatal undersøkelse/screening: Mulighet for å undersøke fosteret i svangerskapet.
- Blod- og urintester: Tester for å påvise lysosomale lagringssykdommer.
- Sykehistorie: Kartlegging av symptomer, tidligere spontanaborter og arvelige disposisjoner.
- Klinisk undersøkelse: Vurdering av avvik fra normal utvikling og typiske sykdomstegn. Legen vil undersøke øyne, hørsel, ansikt, hud, ledd, skjelett, rygg, hjerte, lunger og mageområdet (lever og milt).
- Blodprøver: CRP, SR, hemoglobin, leukocytter, trombocytter, elektrolytter, lever-, nyre- og thyreoideafunksjonsprøver, glukose, kreatin kinase (CK), ANA og urinstix.
- Bildediagnostikk: Ultralyd av mageområdet, røntgen og MR.
- Hjerteundersøkelse: EKG og ekkokardiografi.
- Hørselstest: Audiometri.
- Synstest: Undersøkelse hos øyelege.
- Lungefunksjonstester: For eldre barn.
Diagnosen
Ved mistanke om lagringssykdom tas blodprøver for å undersøke for enzymdefekter. I noen tilfeller gjøres også genetiske analyser.
Behandling
Behandlingsmulighetene varierer for de ulike lagringssykdommene.
- Enzymerstatningsterapi: Den vanligste behandlingsformen. Enzymer gis for å erstatte de som mangler. Brukes blant annet ved Gaucher, Fabry, Pompe og Wolman sykdom, α-mannosidose, NCL type 2, og mukopolysakkaridose type I, II, IV, VI og VII.
- Stamcelletransplantasjon: Benmargstransplantasjon eller transplantasjon av navlestrengsblod.
Behandlingen er ofte livslang og ikke helbredende. Det forskes på genterapi som potensielt kan kurere sykdommene. Dessverre dør fortsatt mange barn med lagringssykdom i løpet av måneder eller år.
Forskjellige typer lagringssykdommer
Lagringssykdommene kan deles inn etter typen molekyler som lagres i kroppen. Hver gruppe har flere undergrupper, og det finnes totalt rundt 50 kjente lagringssykdommer.
Hovedgrupper
- Cystinose (Aminosyredefekt): Redusert transport av aminosyren cystin ut av lysosomene. Den vanligste årsaken til Fanconis syndrom hos barn. Cystinose finnes i tre ulike former, hvorav nyre-type (nefropatisk) starter i løpet av første leveår, intermediær type begynner i tenårene og en voksen-type som bare har øyne-manifestasjoner.
- Glykoproteinlagringssykdom (Mukolipidoser): Opphopning av glykoproteiner og glykolipider. Ligner både mukopolysakkaridoser og sfingolipidoser. Type I (sialidose) forårsakes av mangel på sialidase som kodes av genet NEU1 (6p21.33). Type II (I-celle sykdom) og III (pseudo-Hurler) skyldes mangel på N-acetylglucosaminyl phosphotransferase som kodes av genet GNPTAB (12q23.2). Type IV skyldes mutationer i MCOLN1 genet (locus 19p13.2-13.3) som koder for lysosomal membran kanal og er involvert i Ca2+ signalisering.
- Glykogen lagringssykdom type II (Pompe sykdom): Mangel på enzymet sur α-glukosidase. Glykogen lagres i cellene, spesielt i muskelceller. Fører til muskelsvakhet, hjertesvikt og pustevansker. Sykdommen påvises også blant voksne, til dels på grunn av forsinket diagnostikk. Feilen ligger i GAA genet (17q25.3).
- Lipid (fettstoffer)-lagringssykdommer: Opphopning av fettstoffer. Omfatter blant annet Gaucher’s sykdom, Niemann–Pick sykdom, Gangliosidose og Tay–Sachs sykdom.
- Mukopolysakkaridoser: En gruppe sykdommer der glykosaminoglykaner ikke brytes ned. Fører til skade på skjelett, brusk, sener, nervesystem, øyne, hud og bindevev.
Mukopolysakkaridoser
Definisjon. Mukopolysakkaridoser er en gruppe lagringssykdommer karakterisert ved mangel på eller redusert aktivitet av lysosomale enzymer. Dette fører til opphopning av glykosaminoglykaner i cellene, noe som skader skjelett, brusk, sener, nervesystem, øyne, hud og bindevev. Sykdommene er arvelige og har varierende sykdomsforløp.
Sykdomsforløpet er ulikt mellom de forskjellige typene (se nedenfor): Type I (Hurler sykdom, Scheie), type II (Hunter syndrom), type III (Sanfilippo), type VI (Maro-teaux – Lamy), type VII (Sly) og type IX (hyaluronidasemangler) kjennetegnes alle av stivhet og kontrakturer av ledd. Type IV (Morquio) domineres av hypermobilitet. Mukopolysakkaridose er genetisk forårsaket og skilles fra autoimmune bindevevssykdommer. Sykdomsforløpet er ulikt mellom de forskjellige typene.
Symptomer. De ulike typene mukopolysakkaridose har lignende symptomer, men i ulik grad; Gradvis utvikling i løpet av de første leveårene. Karakteristiske ansiktstrekk med lav neserygg, tykke lepper, stor munn og tunge. Kort overkropp, fortykket hud, stor mage, økt hårvekst på kroppen.
Undersøkelser; Klinisk undersøkelse med fokus på forstørret lever og milt. Urintester for å påvise økt mengde polysakkarider. Enzymanalyser i blodprøver. Prenatal fosterdiagnostikk.
Ulike typer mukopolysakkaridose (MPS type I – IX):
- MPS Type I (Hurler, Hurler-Scheie og Scheies syndrom)
- MPS Type II (Hunters syndrom)
- MPS Type III (Sanfilippo syndrom), deles inn i tye A-D
- MPS Type IV (Morquio syndrom)
- MPS Type V, tilsvarer Type I i form av Scheies syndrom
- MPS Type VI (Maroteaux-Lamy syndrom)
- MPS Type VII (Sly syndrom)
- (MPS, Type VIII er tatt ut av klassifikasjonen fordi den dekkes av de andre tilstandene)
- MPS, Type IX (Hyaluronidase-mangel)
Lignende tilstander (differensialdiagnoser): Mukolipidose med lagring av fettstoffer og et lignende utseende som mukopolysakkaridose. Andre lagringssykdommer (Gaucher, Niemann Pick), Legg-Calve-Pertes sykdom (CLP) som medfører symptomer fra hofter, spondylo-epifyse dysplasi (SED)
Litteratur: Muenzer J, 2011
Andre lagringssykdommer
Det finnes en rekke andre lagringssykdommer, inkludert:
- Fabrys sykdom (mangel på alfa-galactosiderase (a-Gal A))
- Lysosomal sur lipase mangel (LAL-D): Autosomal arvelig lagringssykdom der for lav produksjon av enzymet aktiv lysosomal sur lipase. Symptomer: Fra første leveår ses stor mage, fordøyelsesproblemer, manglende vektøkning, redusert vekst, gul-farget hud. Mildere former diagnostiseres blant unge voksne. Hjertesykdom, leversvikt og slag kan være komplikasjoner. Behandling består av å tilføre enzymet sebelipase-alfa som erstatter manglende enzym. Prenatal fosterdiagnostikk er mulig. Genetisk veiledning (25% risiko for at søsken blir syke). Litteratur: Wierzbicka-Rucińska A, 2016.
- Sykdom i karbohydratmatabilismen (f.eks. glycogen lagringssykdom, G6PD mangel)
- Sykdommer i aminosyremetabolismen (f.eks. fenylketonuri, lønnesirupurinsykdom, glutaric acidemia type 1)
- Sykdommer i urinsyresyklus (f.eks. carbamoyl phosphate synthetase I-mangel)
- Sykdom i organisk syremetabolisme (organic acidurias) (f. eks alkaptonuri, 2-hydroxyglutaric acidurias)
- Sykdom i fettsyreoksydasjon og mitokondriemetabolismen (f. eks medium-chain acyl-coenzyme A dehydrogenase deficiency (MCADD)
- Sykdom i porfyrinmetabolismen ( f. eks. akutt intermitterende porfyri)
- Sykdom i purin- eller pyrimidinmetabolismen (f. eks. Lesch–Nyhan syndrom)
- Sykdom i steroidmetabolismen (f. eks. Lipoid kongenital adrenal hyperplasi, kongenital binyrehyperplasi)
- Sykdom i mitokondrie funksjonen (f. eks Kearns–Sayre syndrom)
- Sykdom i peroxisomal funksjonen ( f. eks Zellweger syndrom)
Litteratur
- Rajkumar V, 2023
- Marques ARA, 2019
- Pastores GM 2010 (Behandling)